Schwannoma: Etiology, Epidemiology, Classification, Clinical Features, Diagnosis, Treatment, and Prognosis
Artemiy G.Neuro-oncological surgeon, MD
19 min read·February 26, 2026
This article is for informational purposes only
The content on this website, including text, graphics, and other materials, is provided for informational purposes only. It is not intended as advice or guidance. Regarding your specific medical condition or treatment, please consult your healthcare provider.
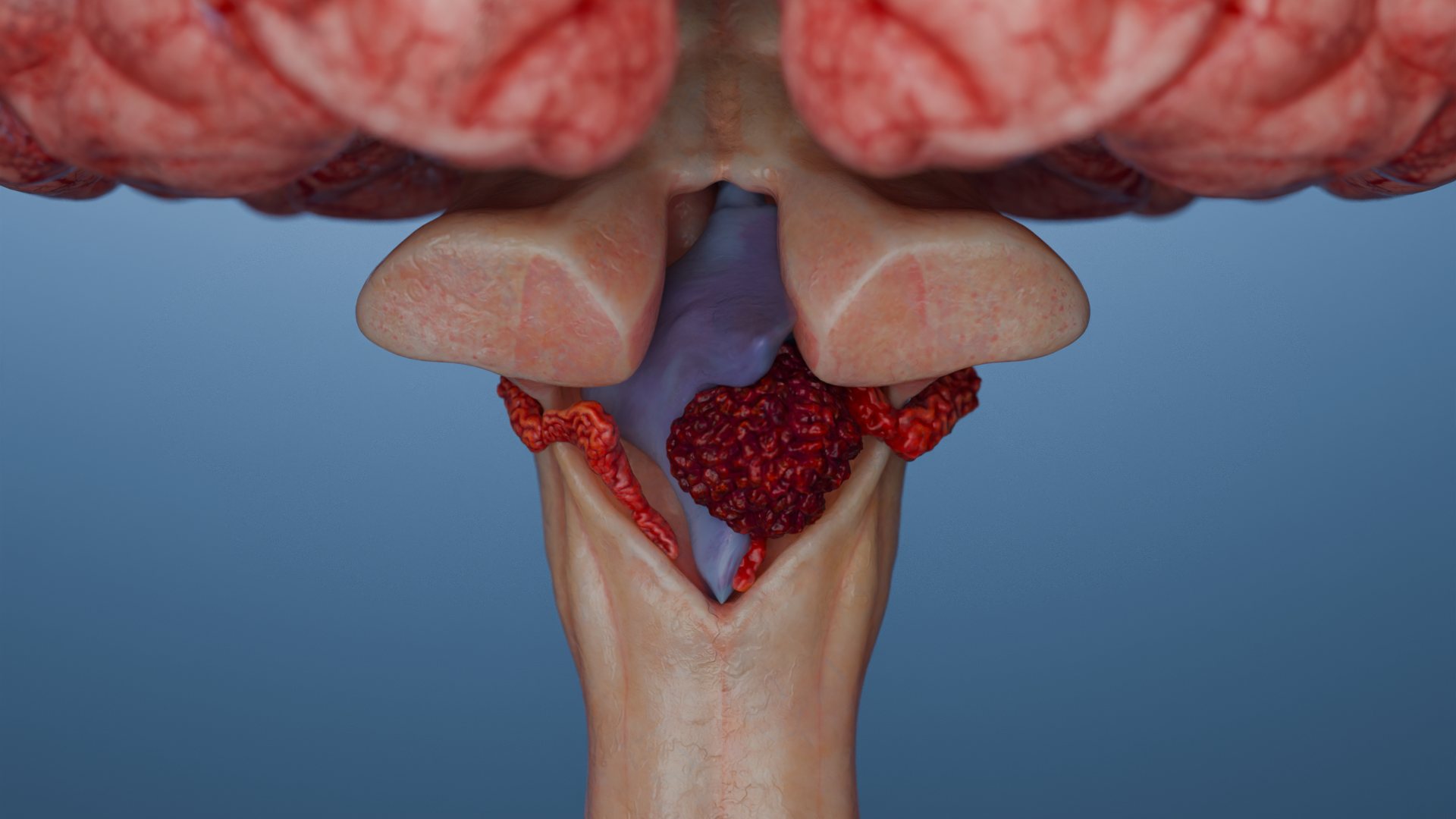
Schwannoma (neurinoma, neurilemmoma) is a typically encapsulated benign tumor arising from Schwann cells in the peripheral and cranial nerves, most often involving the VIII pair (vestibular schwannoma, VS).
Most schwannomas grow slowly, have clear boundaries with the nerve (expansive growth), and very rarely become malignant.
Schwannoma of the spinal nerve (spinal schwannoma)
Schwannoma of the spinal nerve (spinal schwannoma)
Schwannoma of the spinal nerve (spinal schwannoma)
Etiology and molecular background
Sporadic schwannomas are caused by somatic spontaneous mutations in the NF2 gene on chromosome 22q, leading to the loss of merlin protein function, disrupting Schwann cell proliferation control. The pathogenesis of the disease corresponds to the “two-hit” model: the initial NF2 mutation in one allele followed by subsequent loss of heterozygosity or other inactivation of the second allele (LOH 22q, point mutations, mitotic recombination, promoter methylation).
In NF2-related schwannomatosis (formerly termed neurofibromatosis type 2), the mutant NF2 allele is present in the germline. Additional somatic events (“hits”) in NF2 and other genes on chromosome 22 lead to the development of multiple schwannomas (including bilateral vestibular ones), meningiomas, and ependymomas, etc.
In other forms of schwannomatosis (SMARCB1- and LZTR1-associated), mutations in the corresponding tumor suppressor genes on 22q are inherited; additional somatic inactivation of NF2 is necessary for tumor development.
Risk factors for schwannoma development
Presence of NF2-related schwannomatosis: an autosomal dominant syndrome predisposing to schwannomas; pathogenic NF2 variant carriers nearly inevitably develop bilateral vestibular schwannomas and/or multiple other schwannomas.
Presence of SMARCB1/LZTR1-swiftamatosis: heritable syndromes characterized by multiple predominantly non-vestibular schwannomas, in the absence of bilateral vestibular schwannomas.
Neurofibromatosis type 1 (NF1) is not associated with typical schwannomas (NF1 predominantly involves neurofibromas and malignant peripheral nerve sheath tumors, MPNSTs), and schwannomas are not considered a component of NF1.
Previous radiation therapy is a risk factor for the development of MPNSTs, but not of typical schwannomas.
Epidemiology
Schwannomas are the most common benign tumors of peripheral nerves.
Most schwannomas are solitary tumors; the presence of multiple tumors requires exclusion of NF2-related schwannomatosis and other forms of schwannomatosis.
Epidemiology of spinal and peripheral schwannomas
Among intradural extramedullary tumors of the spinal canal, nerve sheath tumors make up about 25%, with approximately 2/3 of these being predominantly sporadic schwannomas.
Only about 2% of all spinal schwannomas occur in patients with NF2; the rest are sporadic incidental findings or symptomatic solitary tumors.
In NF2-related schwannomatosis, spinal tumors (primarily dorsal root schwannomas, but also meningiomas and ependymomas) are identified in 90% of patients during their lifetime, making spinal schwannomas a key epidemiological feature of NF2-related schwannomatosis.
Peripheral (limb, plexus) schwannomas are rare in the general population. A large case series of hundreds of patients shows them as the main pathology among resected benign neoplasms of large peripheral nerve trunks.
Epidemiology of predisposition syndromes (in the context of schwannoma)
NF2-related schwannomatosis: population studies indicate an incidence rate of NF2-related schwannomatosis reaching 1:25000, with a significant proportion being mosaic* forms (at least half of de novo patients).
SMARCB1/LZTR1-related schwannomatosis: data from national neurofibromatosis study centers indicate that this is a much rarer condition than NF2-related schwannomatosis, but with a high individual “burden” of schwannomas in such patients. When analyzing the epidemiology of NF syndromes, schwannomatosis constitutes a minority compared to NF1 and NF2-related schwannomatosis.
Explanation:
*Mosaic forms of schwannomatosis are disease variants where the pathogenic gene variant (typically NF2, SMARCB1, or LZTR1) is present not in all cells of the organism but in some cell clones, arising post-fertilization (post-zygotically). In other words, it is a schwannomatosis caused by somatic mosaicism: some tissues carry the mutation, while others remain genetically normal.
Epidemiology of vestibular schwannomas (VS/acoustic neuroma/schwannoma of the cerebellopontine angle)
Vestibular schwannomas (VS) account for 80–90% of tumors in the cerebellopontine angle in adults; approximately 4–6% are associated with NF2-related schwannomatosis.
Up to 90–95% of patients with NF2 have bilateral vestibular schwannomas by the age of 30, while the vast majority of vestibular schwannomas in the general population are unilateral and sporadic.
In children and young adults with a solitary vestibular schwannoma, the proportion of patients with a germline NF2 mutation is significantly higher than in individuals over the age of 30, making age an important epidemiological marker of latent NF2-related schwannomatosis.
Clinical manifestations of schwannomas
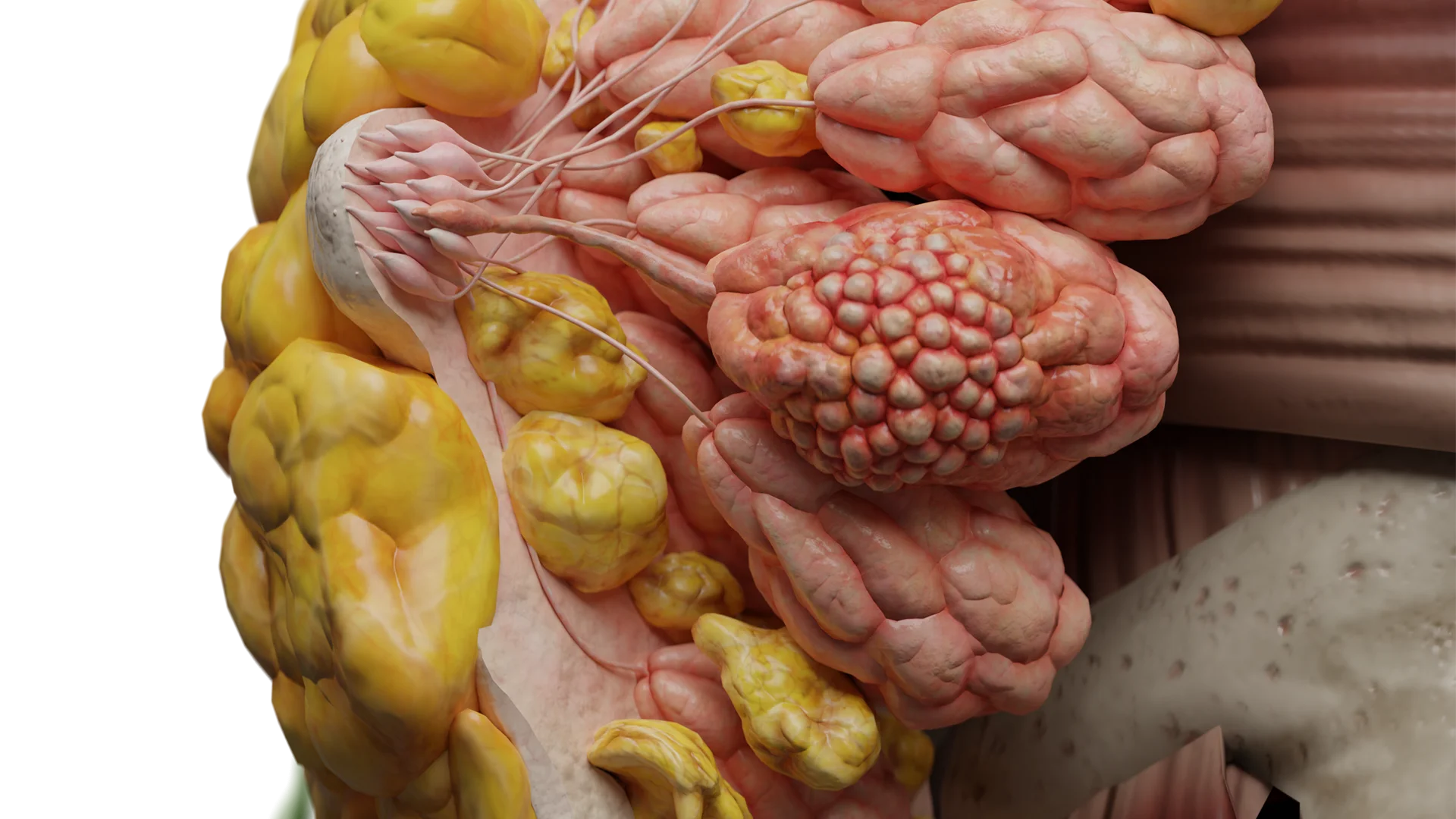
Peripheral schwannomas (of peripheral nerves and plexuses)
Complaints:
Slow-growing painless or painful nodule/tumor along the course of the nerve;
Paresthesias;
Local hypoesthesia or weakness in the nerve distribution area;
Often a positive Tinel’s sign (tingling on tapping) is reported.
Objectively: a firm, elastic formation, mobile across the nerve axis but fixed along its course; neurological deficit is more often sensory, less often motor.
Spinal schwannomas
Spinal schwannomas often arise from dorsal (sensory) roots, remaining asymptomatic for a long time.
They are often an incidental finding during an MRI of the spine, less commonly during an X-ray of the chest.
A variant of spinal schwannoma visualization on chest X-ray investigation
Typical symptoms:
Back pain;
Radicular sensory disturbances;
Motor disorders (muscle weakness below the level of spinal cord compression);
Multiple schwannomas are often accompanied by pain syndrome (often with disabling neuropathic pain in SMARCB1/LZTR1-related schwannomatosis), and in NF2-related schwannomatosis, the disease is often combined with meningiomas and spinal tumors.
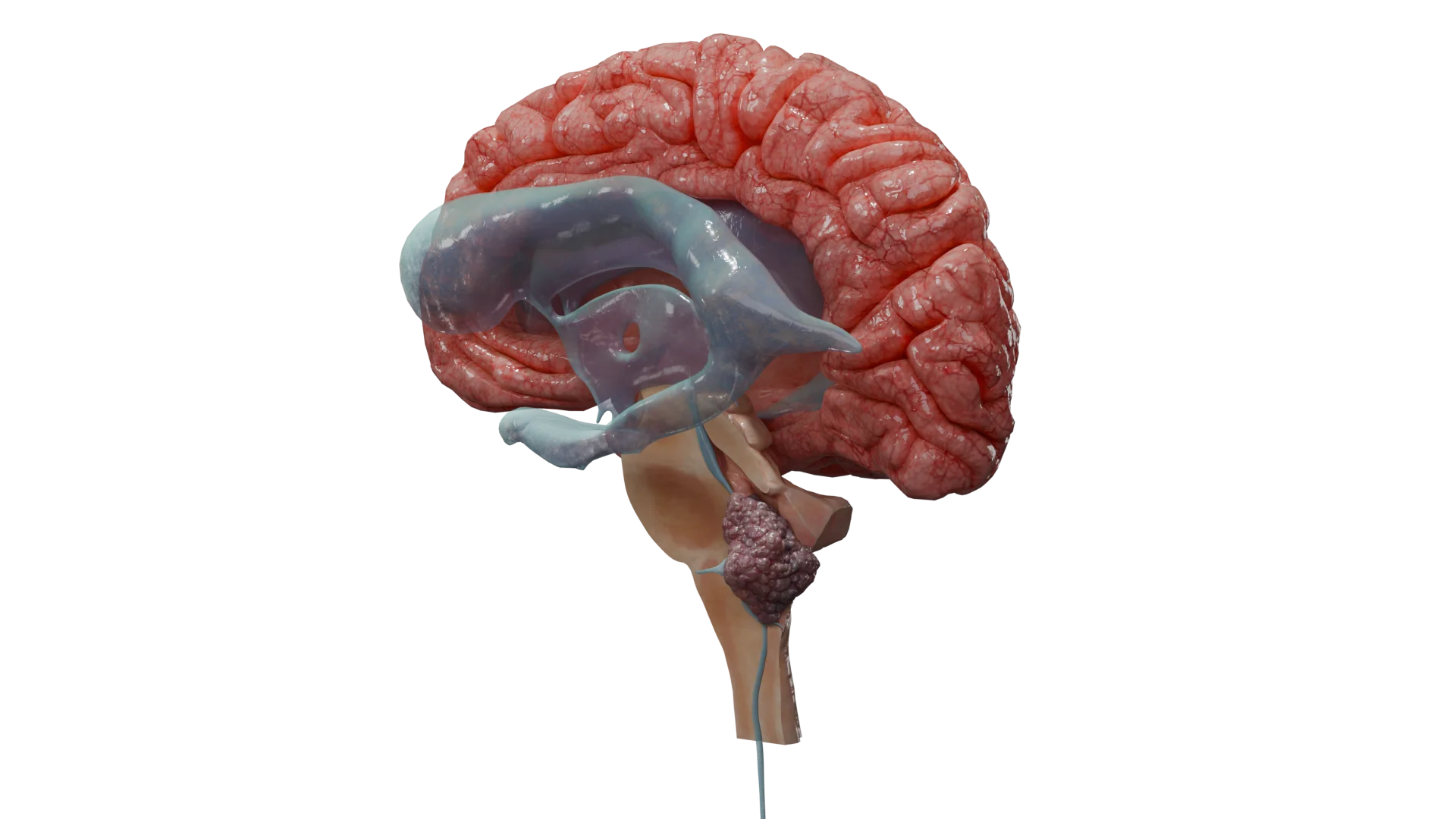
Vestibular (acoustic) neuroma
The specific triad: unilateral (or bilateral in NF2-related schwannomatosis) sensorineural hearing loss, tinnitus (ringing in the ear), and balance disorder.
For larger tumors: headache, ataxia, symptoms of brainstem compression, hydrocephalus, and facial and trigeminal nerve involvement.
Classification of schwannomas
By localization
Peripheral nerves (limbs, plexuses);
Spinal schwannomas (intra/extradural extramedullary). Often (up to 15% of cases) there are “hourglass/dumbbell”-type schwannomas;
Cranial nerves. The most common type is schwannoma of the eighth cranial nerve, also known as “vestibular schwannoma”/“acoustic schwannoma”/“schwannoma of the cerebellopontine angle.” Less commonly, schwannoma of the V (trigeminal) cranial nerve and others are reported.
3D animation: spinal schwannoma of “hourglass”/“dumbbell” type
By cause (based on clinical and genetic characteristics)
Type
Key features
Sporadic schwannoma
Solitary, benign course
NF2-related schwannomatosis
Bilateral vestibular schwannomas + multiple central (spinal) and peripheral schwannomas, meningiomas, ependymomas
SMARCB1/LZTR1-related schwannomatosis
Multiple non-abdominal schwannomas without bilateral vestibular (acoustic), often with significant neuropathic pain syndrome
By histological type
Conventional;
Other types: cellular, plexiform, epithelioid, with degenerative changes (“ancient”).
All histological types of schwannomas mentioned above are benign.
Malignant peripheral nerve sheath tumor (MPNST) and malignant melanotic nerve sheath tumor (MMNST), formerly known as melanotic schwannoma, belong to separate classes of malignant nerve sheath tumors and are not “malignant schwannomas” per WHO 2021 terminology.
For vestibular schwannomas, separate anatomical classifications exist based on tumor localization and size (Koos grading system, Hannover/Samii classification), as well as functional classifications based on hearing (Gardner-Robertson) and facial nerve function (House-Brackmann).
Diagnosis of neurinoma
Clinical presentation and examination
Evaluation of tumor growth dynamics, pain, neurological manifestations; family history of NF1/NF2/schwannomatosis.
In vestibular schwannoma: pure tone audiometry, speech tests, and, if necessary, vestibular tests.
Imaging
MRI with contrast is the method of choice.
Peripheral/spinal: hypointense/isointense on T1, hyperintense on T2, intensely and often homogeneously enhanced with contrast; often have a distinct capsule and eccentric positioning relative to the nerve;
Vestibular: contrast-enhancing tumor within the internal auditory canal extending to the cerebellopontine angle (typical “ice-cream cone sign”).
CT — for assessment of bone structures (expansion of the bony canal or foramina; occasional vertebral destruction).
Morphological verification
For superficial and peripheral tumors, biopsy/total tumor removal with histological examination: Antoni A/B, Verocay bodies, diffuse S100-positivity.
For typical vestibular schwannoma and specific appearance on MRI in immunocompetent patients, biopsy is not required.
Genetic testing
Used not for verification of the tumor itself, but for identification/exclusion of tumor syndromes (NF2-schwannomatosis, SMARCB1/LZTR1-schwannomatosis), assessing family risk, and planning follow-up.
Indications:
Multiple schwannomas;
Young age;
Bilateral vestibular schwannomas associated with another CNS tumor;
Family history.
Differentiation between NF2-schwannomatosis and SMARCB1/LZTR1-schwannomatosis is essential, as it determines follow-up strategy and risk of other tumors.
For solitary sporadic schwannoma in an adult without a family history, testing is usually not required.
Differential diagnosis
Neurofibroma (often in NF1): intraneural, fusiform, non-expansive growth, lack of distinct capsule, presence of axons within the tumor, malignancy risk (MPNST).
Soft tissue tumors: lipoma, desmoid, ganglion, perineurioma, metastatic and other nerve lesions, paraganglioma, sarcoma.
Other intradural extramedullary tumors: meningioma, myxopapillary ependymoma, etc.
In vestibular schwannoma: meningioma of the cerebellopontine angle, epidermoid, metastasis, less commonly — glial tumor.
Treatment of schwannoma
The treatment strategy for schwannomas should be individualized, considering the following:
The goal is a function-preserving total removal; subtotal resection is allowed if necessary to preserve nerve function.
Technique: microscope, intraoperative neuro-monitoring, and identification and preservation of functional roots; in dumbbell tumors — combined approach.
3D animation: spinal schwannoma of “hourglass”/“dumbbell” type
Outcomes: after total resection, recurrences are rare (less than 5%); with residual tumor, continued growth is noted in less than 30% of cases (more often in NF2-schwannomatosis), but the second surgery is required only for a minority.
Vestibular schwannoma
Current guidelines (EANO 2019, systematic reviews) emphasize a strategy based on size, symptoms, and hearing status.
For small/medium tumors with a hearing ear, three strategies are possible:
Observation;
SRS;
Microsurgery with preservation of hearing and facial nerve function.
Systematic reviews show that for small vestibular schwannomas, SRS provides a high rate (>90%) of long-term tumor control and better preservation of facial and auditory nerves compared to surgical treatment.
For large vestibular schwannomas (usually >2.5–3 cm with brainstem compression), primary microneurosurgical resection with decompression of the brainstem is recommended; the retrosigmoid and translabyrinthine approaches are most often used.
The modern trend is “cranial nerve sparing,” that is, subtotal resection with subsequent early SRS on the residual tumor; this “hybrid” approach, according to multicenter series, provides high oncological control and better preservation of VII–VIII nerve function.
For NF2-associated vestibular schwannomas:
Surgical treatment of NF2-associated vestibular schwannomas is complicated by their multifocal nature and low vascularization, as well as a high risk of bilateral deafness.
Whenever possible, preference is given to less aggressive, hearing-preserving, or combined interventions, considering the potential effect of drug therapy (bevacizumab).
Radiotherapy
Stereotactic radiosurgery and radiotherapy (SRS&SRT)
Indications for peripheral/spinal schwannomas include:
Recurrence/residual tumor after surgery;
Inability to perform surgical treatment.
Stereotactic radiotherapy demonstrates high local control (>90%) and a low risk of complications in spinal schwannomas.
For vestibular schwannoma:
SRS is an option for small/medium vestibular schwannomas, especially in patients where preserving facial and auditory nerve function is the primary goal with minimal intervention;
Growth control >90% over a 10–15 year follow-up period with a single 11–13 Gy dose.
The risk of late hearing deterioration persists but is generally lower than after microsurgery in relevant cohorts.
Radiotherapy in NF2
Used more cautiously due to a higher risk of secondary neoplasms (MPNST, sarcomas, meningiomas) in patients with tumor syndromes.
In NF2, preference is given to surgery and/or targeted therapy (bevacizumab); radiotherapy is used if surgery is not feasible or seems dangerous.
Medication therapy
For sporadic isolated schwannomas (peripheral or spinal), there is no convincing evidence of the benefit of systemic therapy. The standard treatment is surgery/observation; radiotherapy is used if necessary.
The only agent with a solid evidence base and real clinical utility in NF2-schwannomatosis is bevacizumab for progressing, functionally significant vestibular schwannomas (and partially other NF2-tumors).
Bevacizumab
Anti-VEGF monoclonal antibody is the main proven systemic option in progressing NF2-associated vestibular schwannomas threatening hearing or brainstem compression.
A systematic meta-analysis of 8 cohorts (161 patients) showed a partial response in tumor volume in ≈41%, stabilization in 47%, and progression in 7%; hearing improvement in about 20% and stabilization (no worsening of hearing) in 69% of patients.
Limitations of bevacizumab therapy:
Proteinuria;
Hypertension;
Risk of serious vascular complications (including rare intracranial hemorrhages).
Long-term safety and prolonged efficacy remain subjects of study.
Other targeted and experimental approaches
mTOR inhibitor everolimus: activity in NF2-associated vestibular schwannomas was found to be limited and inconsistent.
Tyrosine kinase inhibitors (lapatinib, EGFR/ErbB inhibitors) demonstrated single partial responses in small studies in patients with NF2.
Trials of the ALK inhibitor brigatinib and HDAC inhibitors (e.g., AR-42) are ongoing for NF2-associated tumors; currently, these are experimental options within clinical studies.
Prognosis of the disease
Sporadic benign schwannomas (peripheral, spinal, vestibular) with adequate treatment rarely relapse, practically do not affect overall survival, and allow preservation or improvement of neurological status in most patients.
After total resection of spinal schwannoma, recurrences are observed in less than 5%.
In NF2-schwannomatosis, the prognosis is determined by the multiplicity of tumors (vestibular schwannomas, meningiomas, ependymomas), progressive neurosensory deafness, balance disorders, and polyneuropathy. Despite improvements in diagnosis and therapy, life expectancy remains decreased compared to the general population.
Malignant transformation of a typical schwannoma is extremely rare. MPNST generally arises from plexiform neurofibromas in NF1 or after radiotherapy and has a significantly worse prognosis (5-year survival of 34–60%).
The “wait and scan” observation tactic for asymptomatic schwannomas:
Justified and widely accepted for sporadic small vestibular and peripheral/spinal schwannomas in the absence of critical structure compression and good patient compliance;
Requires a clear protocol of MRI monitoring (at least two early studies to determine growth type, then the interval is personalized);
Must be accompanied by detailed patient information regarding the risk of growth and the possible necessity for subsequent interventions.
Schwannoma growth rate
The average growth of most schwannomas (including vestibular) is negligible (≈1 mm/year in diameter or modest volume increase), yet there is an infrequent subset of rapidly growing tumors that must be promptly identified.
Peripheral and spinal schwannomas
Most peripheral and spinal schwannomas grow slowly: about 5–10% of the volume per year.
In the study by El Sayed L et al. in a case series of 47 peripheral schwannomas (sporadic and schwannomatosis-related), the relative annual growth was approximately 34% per year; meanwhile, ≈19% of tumors did not grow or shrank, and ≈13% exhibited “rapid” growth (>2 cm³/year and >35%/year).
In the study by Lubelski D. et al. in a case series of 227 patients investigating the natural course of cervical plexus, peripheral, and spinal tumors, growth subtypes were identified: “slow” growth (~5–10% of the volume per year), “moderate,” and “rapid” (>80% per year), with growth type often determined after two follow-up evaluations.
3D animation: growth of spinal extradural schwannoma
Vestibular schwannomas (VS)
A systematic review by Yoshimoto Y. of natural history showed:
Growth frequency (any positive increase) is ≈46% over an average follow-up period of 38 months;
Regression: 8%;
The average growth rate by maximum diameter is approximately 1.2 mm/year.
A modern review by Paldor I. et al. reports similar figures for VS growth: average growth of 0.99–1.11 mm/year; if the tumor has already shown growth by the first follow-up MRI, the expected growth rate is ≈3 mm/year, with rapid growth factors (>4 mm/year) including cysts, hemorrhages, and hormonal therapy.
The range of speeds is broad: from no growth or even reduction to 6–17 mm/year in individual case series.
Find more scientifically accurate content on our social media
Subscribe and don’t miss out the latest resources
Key practical focus for the physician
Any rapid change in size, pain character, or neurological status in a patient with a known schwannoma (especially with NF1) requires exclusion of MPNST (MRI with DWI, PET-CT, biopsy).
In young patients with vestibular schwannoma, NF2-related schwannomatosis should be actively excluded (MRI of the entire CNS, genetic testing).
For asymptomatic patients with small schwannomas and a stable clinical and radiographic picture, an observation strategy with regular MRI is safe.
FAQ
1. What is a schwannoma, and is it cancerous?
A schwannoma (or neurinoma) is a benign tumor that develops from the Schwann cells of the sheaths of peripheral or cranial nerves. It is not cancer, has a capsule, is characterized by slow expansive growth, and very rarely progresses to malignancy (in other words, shows rare transformation into a malignant form).
2. What are the causes of neurinoma?
The etiology of the disease is associated with mutations of the NF2 gene on chromosome 22, leading to the loss of the function of the merlin protein and a disruption in cell division control. These mutations can occur spontaneously (sporadic cases) or be inherited within genetic syndromes, for example, NF2-schwannomatosis (previously known as neurofibromatosis type 2) or other forms of schwannomatosis.
3. What symptoms does vestibular (acoustic) neurinoma cause?
The tumor of the VIII pair of cranial nerves is characterized by a specific clinical triad: unilateral sensorineural hearing loss, tinnitus (ringing in the ear), and balance disorder.
4. How dangerous is schwannoma, and is associated mortality high?
The prognosis for this disease is favorable because the tumor is benign and, in most cases, grows slowly, so the direct mortality is extremely low.
5. What is cauda equina neurinoma, and what are its symptoms?
This is a schwannoma located in the lumbosacral spine, where the nerve roots pass. Symptoms include back pain, radicular sensory disturbances in the legs, sometimes muscle weakness in the affected leg, and pelvic organ dysfunction.
6. Is schwannoma visible on CT or ultrasound?
MRI: Primary diagnostic tool. On ultrasound, only superficial schwannomas of the peripheral nerves of the limbs or neck are visible; this method is not suitable for deep tumors. Computed tomography (CT) visualizes soft tissues worse but is important for assessing bone changes: ear canal expansion, intervertebral foramina, or vertebral destruction.
7. What is Morton’s neuroma of the foot?
Morton’s neuroma is not a true tumor of Schwann cells but a localized thickening (fibrosis) of the plantar nerve sheath of the foot due to chronic compression. Unlike spinal or cranial schwannomas, it manifests as burning pain between the toes while walking and carries no oncological risks.
8. What happens to schwannoma after surgery?
With total surgical removal of the tumor, relapses occur very rarely — in less than 5% of cases. If part of the capsule was left to preserve nerve function, continued growth of residual tissue is possible, observed in less than 30% of patients.
References
1.
VOKA 3D Anatomy & Pathology – Complete Anatomy and Pathology 3D Atlas [Internet]. VOKA 3D Anatomy & Pathology.
Available from: https://catalog.voka.io/
2.
Goldbrunner R, et al. EANO guideline on the diagnosis and treatment of vestibular schwannoma. Neuro-Oncology. 2019;21(3):e8–e23.
3.
CNS Guidelines Committee. Evidence-based guidelines for the treatment of adults with vestibular schwannoma (update 2025). Congress of Neurological Surgeons.
4.
Sergi B, et al. Management of vestibular schwannoma: a systematic review with focus on radiotherapy vs surgery vs wait-and-scan. Journal of Personalized Medicine. 2022;12:1616.
5.
Pontillo V, et al. Hearing preservation surgery for vestibular schwannoma: systematic review. 2024.
6.
Laufer I, Bilsky M. Intradural nerve sheath tumors. UpToDate, 2023 update.
Evans DG, et al. NF2-related schwannomatosis. UpToDate, 2022 update.
9.
Evans DG, Mostaccioli S, Pang D, et al. ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis. Eur J Hum Genet. 2022;30(7):812–817.
10.
Lu VM, Ravindran K, Graffeo CS, et al. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: a systematic review and meta-analysis of treatment outcomes. J Neurooncol. 2019;144(2):239-248.
11.
EANS Task Force. Surgical management of large vestibular schwannomas: systematic review and recommendations. Acta Neurochir. 2020.
12.
Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967–1977.
13.
Lubelski D, Pennington Z, Ochuba A, et al. Natural History of Brachial Plexus, Peripheral Nerve, and Spinal Schwannomas. Neurosurgery. 2022;91(6):883–891.
14.
El Sayed L, Masmejean EH, Parfait B, Kalamarides M, Biau D, Peyre M. Natural history of peripheral nerve schwannomas. Acta Neurochir (Wien). 2020;162(8):1883-1889.
15.
Yoshimoto Y. Systematic review of the natural history of vestibular schwannoma. J Neurosurg. 2005;103(1):59–63. doi:10.3171/jns.2005.103.1.0059
16.
Paldor I, Chen AS, Kaye AH. Growth rate of vestibular schwannoma. J Clin Neurosci. 2016;32:1–8. doi:10.1016/j.jocn.2016.05.003
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Thank you!
Your message is sent! Our experts will contact you shortly. If you have any additional questions, please contact us at info@voka.io
Cookie Consent
We use cookies to enhance your browsing experience, analyze site traffic, and deliver content. Please choose whether you accept all cookies or wish to reject non-essential tracking.
Cookie Preferences
Manage your cookie preferences below:
Essential cookies enable basic functions and are necessary for the proper function of the website.
Name
Description
Duration
Geolocation Config
This cookie is used to store the consent settings based on the visitor's location.
30 days
Cookie Preferences
This cookie is used to store the user's cookie consent preferences.
30 days
Google reCAPTCHA helps protect websites from spam and abuse by verifying user interactions through challenges.
Name
Description
Duration
_GRECAPTCHA
Google reCAPTCHA sets a necessary cookie (_GRECAPTCHA) when executed for the purpose of providing its risk analysis.
179 days
Statistics cookies collect information anonymously. This information helps us understand how visitors use our website.
Google Analytics is a powerful tool that tracks and analyzes website traffic for informed marketing decisions.
ID used to identify users for 24 hours after last activity
24 hours
_gat
Used to monitor number of Google Analytics server requests when using Google Tag Manager
1 minute
_gac_
Contains information related to marketing campaigns of the user. These are shared with Google AdWords / Google Ads when the Google Ads and Google Analytics accounts are linked together.
90 days
__utma
ID used to identify users and sessions
2 years after last activity
__utmt
Used to monitor number of Google Analytics server requests
10 minutes
__utmb
Used to distinguish new sessions and visits. This cookie is set when the GA.js javascript library is loaded and there is no existing __utmb cookie. The cookie is updated every time data is sent to the Google Analytics server.
30 minutes after last activity
__utmc
Used only with old Urchin versions of Google Analytics and not with GA.js. Was used to distinguish between new sessions and visits at the end of a session.
End of session (browser)
__utmz
Contains information about the traffic source or campaign that directed user to the website. The cookie is set when the GA.js javascript is loaded and updated when data is sent to the Google Anaytics server
6 months after last activity
__utmv
Contains custom information set by the web developer via the _setCustomVar method in Google Analytics. This cookie is updated every time new data is sent to the Google Analytics server.
2 years after last activity
__utmx
Used to determine whether a user is included in an A / B or Multivariate test.
18 months
_ga
ID used to identify users
2 years
_gali
Used by Google Analytics to determine which links on a page are being clicked
30 seconds
Clarity is a web analytics service that tracks and reports website traffic.