Schwannom: Ätiologie, Epidemiologie, Klassifikation, Klinik, Diagnostik, Therapie und Prognose
Gontsov A.Neuro-onkologischer Chirurg, MD
19 min lesen·Februar 26, 2026
Dieser Artikel dient nur zu Informationszwecken
Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
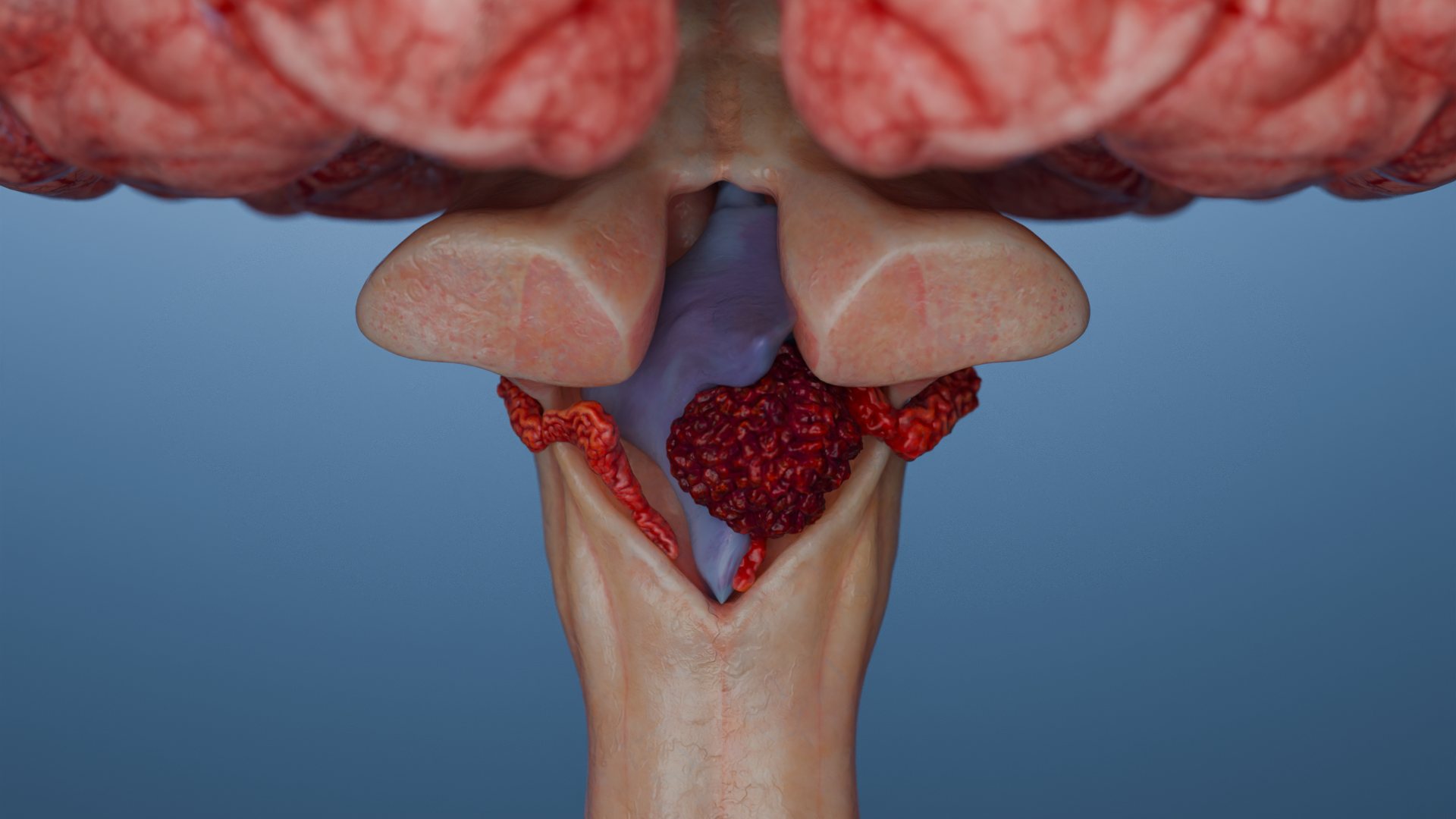
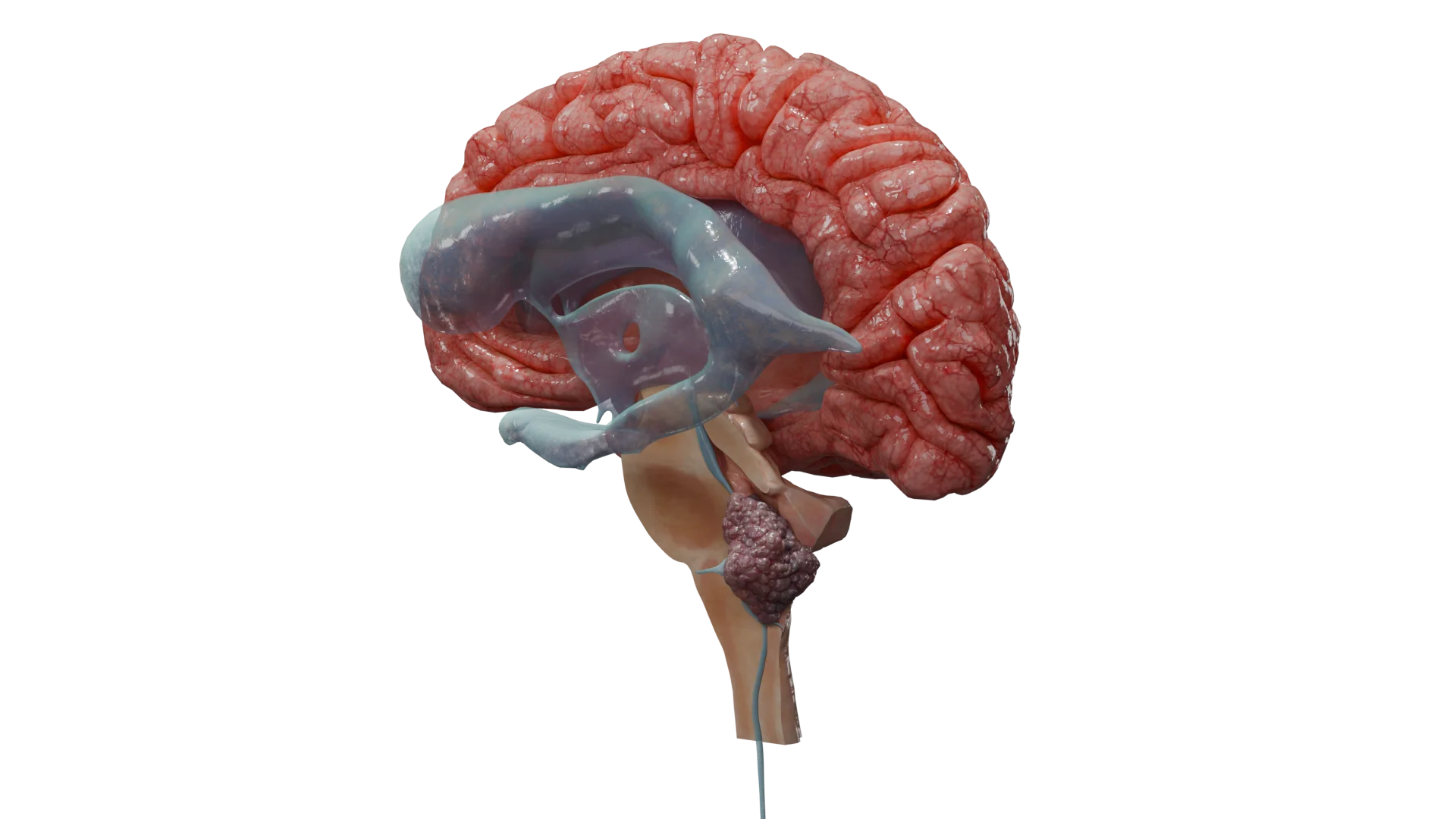
Als Schwannom (Neurinom, Neurilemmom, Schwann-Zell-Tumor) bezeichnet man einen gutartigen, in der Regel eingekapselten Tumor, der von den Schwann-Zellen der peripheren und Hirnnerven, meist der 8. Hirnnerven (vestibuläres Schwannom, Akustikusneurinom, kurz VS, AKN oder AN), ausgeht.
Die meisten Schwannome wachsen langsam, weisen klare Grenzen zum Nerv auf (expansives Wachstum) und werden äußerst selten bösartig.
Sporadische Schwannome resultieren aus somatischen spontanen Mutationen des NF2-Gens auf Chromosom 22q, die zu einem Funktionsverlust des Merlin-Proteins führen, wodurch die Kontrolle der Proliferation der Schwann-Zellen gestört wird. Die Pathogenese der Erkrankung entspricht dem „Zwei-Treffer-Modell“ (two-hit): eine initiale NF2-Mutation in einem Allel und ein anschließender Verlust der Heterozygotie oder eine andere Inaktivierung des zweiten Allels (LOH 22q, Punktmutationen, mitotische Rekombination, Promotor-Methylierung).
Bei der NF2-bedingten Schwannomatose (früher Neurofibromatose Typ 2 genannt) liegt das mutierte NF2-Allel in der Keimbahn vor. Zusätzliche somatische Ereignisse („Hit“ bzw. „Treffer“) in NF2 und anderen Genen des Chromosoms 22 führen zur Entwicklung multipler Schwannome (einschließlich bilateraler vestibulärer Schwannome), Meningeome, Ependymome und anderer Tumoren.
Bei anderen Formen der Schwannomatose (SMARCB1- und LZTR1-assoziierte Schwannomatose) werden Mutationen der gleichnamigen Suppressorgene auf 22q vererbt. Für die Tumorentwicklung ist eine zusätzliche somatische Inaktivierung von NF2 erforderlich.
Risikofaktoren für Schwannome
Vorliegen einer NF2-Schwannomatose: Es handelt sich um ein autosomal-dominantes Syndrom mit einer Veranlagung für Schwannome; bei Trägern einer pathogenen NF2-Variante entstehen fast zwangsläufig bilaterale vestibuläre Schwannome und/oder zahlreiche andere Schwannome.
Vorliegen einer SMARCB1/LZTR1-Schwannomatose: Dabei geht es um erbliche Syndrome, die durch multiple, überwiegend nicht vestibuläre Schwannome gekennzeichnet sind, wobei bilaterale vestibuläre Schwannome fehlen.
Neurofibromatose Typ 1 (NF1) ist nicht mit klassischen Schwannomen assoziiert (bei NF1 überwiegen Neurofibrome und MPNST), und Schwannome werden nicht als Bestandteil von NF1 angesehen.
Eine vorangegangene Strahlentherapie ist ein Risikofaktor für das Auftreten von MPNST, jedoch nicht für typische Schwannome.
Epidemiologie
Schwannome sind die häufigsten gutartigen Tumoren der peripheren Nerven.
Die meisten Schwannome sind solitäre Tumoren; multiple Raumforderungen erfordern den Ausschluss von NF2-Schwannomatose sowie anderen Formen der Schwannomatose.
Epidemiologie von spinalen und peripheren Schwannomen
Unter den intraduralen extramedullären Tumoren des Wirbelkanals machen Nervenscheidentumoren etwa 25 % aus, davon sind etwa zwei Drittel Schwannome, die überwiegend sporadisch auftreten.
Nur etwa 2 % aller spinalen Schwannome treten bei Patienten mit NF2 auf; die übrigen sind sporadische Zufallsbefunde oder symptomatische Einzeltumoren.
Bei NF2-Schwannomatose werden spinale Tumoren (überwiegend Schwannome der dorsalen Wurzeln, aber auch Meningeome und Ependymome) bei 90 % der Patienten im Laufe ihres Lebens festgestellt, was spinale Schwannome zu einem der wichtigsten epidemiologischen Merkmale der NF2-Schwannomatose macht.
Periphere (im Bereich der Extremitäten und Nervengeflechte) Schwannome sind in der Allgemeinbevölkerung selten. Eine große Serie mit Hunderten von Patienten zeigt sie als Hauptnosologie unter den resezierten gutartigen Neubildungen der peripheren Nerven der großen Stämme.
Epidemiologie der Prädispositionssyndrome (im Kontext von Schwannomen)
NF2-Schwannomatose: In populationsbasierten Studien erreicht die Häufigkeit der NF2-Schwannomatose 1:25.000, wobei ein erheblicher Teil der Fälle durch Mosaikformen* repräsentiert wird (mindestens die Hälfte der De-novo-Patienten).
SMARCB1/LZTR1-Schwannomatose: Daten aus nationalen Registern von Zentren zur Erforschung der Neurofibromatose zeigen, dass dies eine erheblich seltener vorkommende Erkrankung ist als NF2-Schwannomatose, jedoch mit einer hohen individuellen „Belastung“ durch Schwannome bei diesen Patienten. Bei der Analyse der Epidemiologie der NF-Syndrome stellt die Schwannomatose im Vergleich zur NF1- und NF2-Schwannomatose eine Minderheit dar.
Erläuterung:
* Mosaikformen der Schwannomatose sind Krankheitsvarianten, bei denen die pathogene Genvariante (in der Regel NF2, SMARCB1 oder LZTR1) nicht in allen Zellen des Organismus, sondern nur in einem Teil der nach der Befruchtung (postzygotisch) entstandenen Zellklone vorhanden ist. Mit anderen Worten handelt es sich um eine durch somatischen Mosaizismus bedingte Schwannomatose: Ein Teil des Gewebes weist eine Mutation auf, während der andere Teil genetisch normal bleibt.
Epidemiologie vestibulärer Schwannome (Vestibularisschwannome / Akustikusneurinome / VS / AKN / AN / Schwannome im Bereich des Kleinhirnbrückenwinkels)
Vestibuläre Schwannome (VS) machen 80–90 % der Tumoren im Bereich des Kleinhirnbrückenwinkels bei Erwachsenen aus; etwa 4–6 % sind mit NF2-Schwannomatose assoziiert.
Bis zu 90–95 % der Patienten mit NF2 weisen im Alter von 30 Jahren bilaterale vestibuläre Schwannome auf, während die überwiegende Mehrheit der vestibulären Schwannome in der Allgemeinbevölkerung einseitig und sporadisch auftritt.
Bei Kindern und jungen Erwachsenen mit einem einzelnen vestibulären Schwannom ist der Anteil der Patienten mit einer Keimbahn-NF2-Mutation deutlich höher als bei Personen über 30 Jahren, weshalb das Alter ein wichtiger epidemiologischer Marker für eine verborgene NF2-Schwannomatose ist.
Klinische Manifestationen von Schwannomen
Periphere Schwannome (Schwannome der peripheren Nerven und Nervengeflechte)
Beschwerden:
Langsam wachsender schmerzloser oder schmerzhafter Knoten/Tumor entlang des Nervs
Parästhesien
Lokale Hypästhesie oder Schwäche im Innervationsbereich
Häufig: ausgeprägtes Hoffmann-Tinel-Zeichen (Parästhesien beim Beklopfen)
Objektiv: Erkennbar ist eine dichte, elastische Struktur, die quer zur Nervenachse beweglich, aber entlang des Nervenverlaufs fixiert ist; neurologische Defizite sind häufiger sensorisch als motorisch.
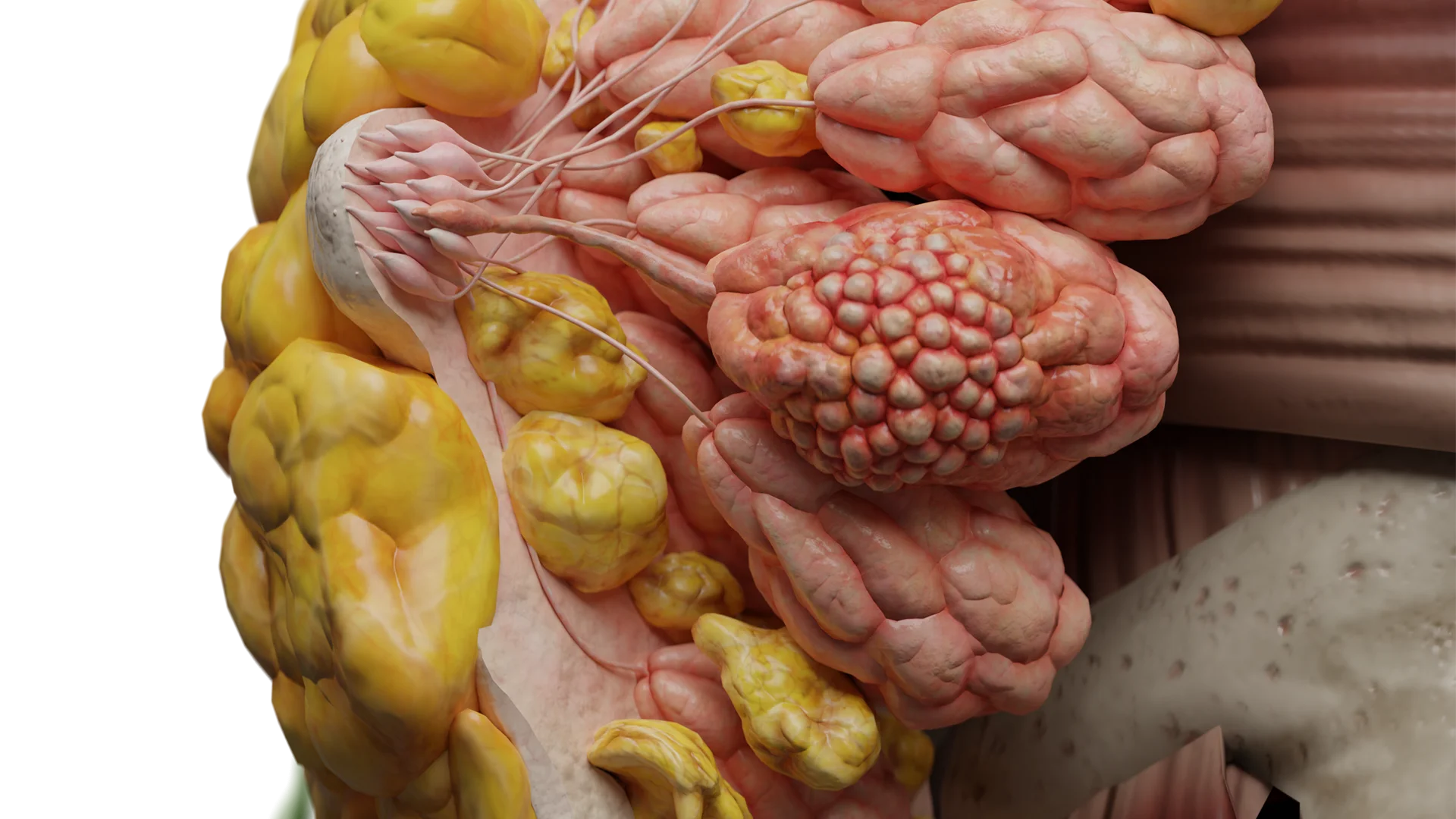
Spinale Schwannome
Spinale Schwannome gehen häufig von den dorsalen (sensiblen) Wurzeln aus und verlaufen lange asymptomatisch.
Sie sind häufig ein Zufallsbefund bei einer MRT-Untersuchung der Wirbelsäule, seltener bei einer Röntgenuntersuchung des Brustkorbs.
Variante der Visualisierung eines spinalen Schwannoms bei einer Röntgenuntersuchung des Brustkorbs
Typische Symptome:
Rückenschmerzen
Radikuläre Empfindungsstörungen
Motorische Störungen (Muskelschwäche unterhalb des Bereichs der Rückenmarkskompression)
Multiple Schwannome gehen häufig mit Schmerzen einher (oft mit behindernden neuropathischen Schmerzen bei SMARCB1/LZTR1-Schwannomatose), und bei der NF2-Schwannomatose tritt die Erkrankung nicht selten zusammen mit Meningeomen und Rückenmarkstumoren auf.
Bei großen Tumoren: Kopfschmerzen, Ataxie, Symptome der Hirnstammkompression und Hydrozephalus, Läsion des N. facialis und N. trigeminus.
Klassifikation der Schwannome
Nach Lokalisation
Schwannome der peripheren Nerven (Extremitäten, Nervengeflechte)
Spinale Schwannome (intra-/extradural extramedullär): häufig (bis zu 15 % der Fälle) Schwannome vom Typ „Sanduhrgeschwulst“ / „hantelförmiger Tumor“ vorkommend
Schwannome der Hirnnerven: am häufigsten Schwannome des 8. Hirnnervs, auch vestibuläre Schwannome, Akustikusneurinome, Schwannome im Bereich des Kleinhirnbrückenwinkels genannt; weniger häufig Schwannome des 5. Nervs (N. trigeminus) u. a. vorkommend
3D-Animation: spinales Schwannom vom Typ „Sanduhrgeschwulst“ / „hantelförmiger Tumor“
Nach Ursache (im Rahmen von klinisch-genetischen Merkmalen)
Multiple nicht abdominelle Schwannome ohne bilaterale vestibuläre Schwannome (Akustikusneurinome), häufig mit ausgeprägtem neuropathischem Schmerzsyndrom
Nach histologischem Typ
Klassisch
Weitere Typen: zellulär, plexiform, epitheloid, mit degenerativen Veränderungen („ancient“)
Alle oben genannten histologischen Typen von Schwannomen sind gutartig.
Maligne periphere Nervenscheidentumoren (MPNST) und maligne melanotische Nervenscheidentumoren (MMNST), die zuvor als melanotische Schwannome bezeichnet wurden, gehören zu separaten Klassen von malignen Nervenscheidentumoren und gelten nicht als „maligne Schwannome“ gemäß WHO-Terminologie 2021.
Für vestibuläre Schwannome gibt es verschiedene anatomische Klassifikationen, die sich nach der Lokalisation und der Größe des Tumors richten (Koos-Klassifikation, Hannover-/Samii-Klassifikation), sowie funktionelle Klassifikationen, die sich am Hörvermögen (Gardner-Robertson-Klassifikation) und an der Funktion des Gesichtsnervs (House-Brackmann-Klassifikation) orientieren.
Diagnostik von Neurinomen
Symptome und Untersuchung
Dazu gehören Bewertung des Tumorwachstums, der Schmerzen, der neurologischen Erscheinungen; NF1-/NF2-Schwannomatose in der Familienanamnese.
Bei vestibulären Schwannomen werden Tonaudiometrie, Sprachtests, bei Bedarf Gleichgewichtstests durchgeführt.
Bildgebung
Als Methode der Wahl gilt MRT mit Kontrastmittel.
Peripher/spinal: hypo- / isointens in der T1-Sequenz, hyperintens in der T2-Sequenz, intensiv und häufig homogen kontrastmittelanreichernd; häufig mit deutlich erkennbarer Kapsel und exzentrischer Lage zum Nerv
Vestibulär: kontrastmittelanreichernder Tumor im inneren Gehörgang mit Ausbreitung in den Kleinhirnbrückenwinkel (klassisches „Ice-Cream-Cone-Sign“)
CT wird zur Beurteilung der Knochenstrukturen eingesetzt (Erweiterung des Knochenkanals und des Zwischenwirbellochs; gelegentlich treten Zerstörungen der Wirbel auf).
Morphologische Verifizierung
Bei oberflächlichen und peripheren Tumoren erfolgt eine Biopsie bzw. eine vollständige Tumorentfernung mit histologischer Untersuchung: Antoni A/B, Verokai-Körperchen, diffuse S100-Positivität.
Bei einem typischen vestibulären Schwannom und einem klassischen MRT-Befund ist bei immunkompetenten Patienten keine Biopsie erforderlich.
Gentests
Gentests dienen nicht zur Bestätigung des Tumors selbst, sondern zur Erkennung bzw. zum Ausschluss eines Tumorsyndroms (NF2-Schwannomatose, SMARCB1/LZTR1-Schwannomatose), zur Einschätzung des Risikos für die Familie und zur Planung der Verlaufskontrolle.
Indikationen:
Multiple Schwannome
Junges Alter
Bilaterale vestibuläre Schwannome, kombiniert mit einem anderen ZNS-Tumor
Familienanamnese
Eine Differenzierung zwischen NF2-Schwannomatose und SMARCB1/LZTR1-Schwannomatose ist zwingend erforderlich, da dies die Verlaufskontrolle und das Risiko anderer Tumoren bestimmt.
Bei einem einzelnen sporadischen Schwannom bei Erwachsenen ohne familiäre Vorbelastung ist normalerweise kein Gentest erforderlich.
Differentialdiagnose
Neurofibrome (häufig bei NF1): intraneural, fusiform, nicht expansives Wachstum, Fehlen einer erkennbaren Kapsel, Vorliegen von Axonen im Tumorinneren, Risiko einer Malignisierung (MPNST).
Das Ziel ist eine funktionserhaltende vollständige Entfernung. Bei Bedarf ist eine subtotale Resektion zulässig, um die Nervenfunktion zu erhalten.
Verfahrenstechnik: Mikroskop, intraoperatives Neuromonitoring, Identifizierung und Erhaltung der funktionellen Nervenwurzeln; kombinierter Zugang bei hantelförmigen Tumoren.
3D-Animation: spinales Schwannom vom Typ „Sanduhrgeschwulst“ / „hantelförmiger Tumor“
Ergebnisse: Nach einer totalen Resektion sind Rezidive selten (weniger als 5 %); bei Residualtumoren (Resttumoren) ist in weniger als 30 % der Fälle ein weiteres Wachstum erkennbar (bei NF2-Schwannomose häufiger), doch ist eine erneute Operation nur bei einer Minderheit erforderlich.
Vestibuläres Schwannom
Aktuelle Empfehlungen (EANO 2019, systematische Übersichtsarbeiten) betonen eine Strategie, die sich nach Größe, Symptomen und Hörstatus richtet.
Bei kleinen/mittleren Tumoren mit intaktem Gehör kommen drei Strategien in Frage:
Überwachung
SRS
Mikrochirurgie unter Erhaltung des Gehörs und der Funktion des Gesichtsnervs
Systematische Übersichtsarbeiten zeigen, dass bei kleinen vestibulären Schwannomen die SRS im Vergleich zur chirurgischen Behandlung eine hohe Rate (> 90 %) an langfristiger Tumorkontrolle sowie bessere Ergebnisse hinsichtlich der Erhaltung des Gesichts- und Hörnervs gewährleistet.
Bei großen vestibulären Schwannomen (in der Regel > 2,5–3 cm, mit Kompression des Hirnstamms) wird eine primäre mikrochirurgische Resektion mit Hirnstammdekompression empfohlen. Am häufigsten werden der retrosigmoidale und der translabyrinthäre Zugang eingesetzt.
Der aktuelle Trend ist „Cranial Nerve Sparing“ („Schonung des Hirnnervs“), d. h. eine subtotale Resektion mit anschließender frühzeitiger SRS des Residualtumors. Dieser „Hybridansatz“ gewährleistet laut Daten aus multizentrischen Studien eine hohe onkologische Kontrolle und bessere Ergebnisse hinsichtlich der Erhaltung der Funktion der 7.–8. Hirnnerven.
Bei NF2-assoziierten vestibulären Schwannomen:
Die chirurgische Behandlung von NF2-assoziierten vestibulären Schwannomen wird durch deren multifokalen Charakter und geringe Durchblutung sowie durch das hohe Risiko einer beidseitigen Taubheit erschwert.
Nach Möglichkeit werden weniger aggressive, hörerhaltende oder kombinierte Eingriffe bevorzugt, wobei die mögliche Wirkung einer medikamentösen Therapie (Bevacizumab) berücksichtigt wird.
Strahlentherapie
Stereotaktische Radiochirurgie (SRS) und Strahlentherapie (SRT)
Für periphere/spinale Schwannome gelten folgende Indikationen:
Rezidiv/Residualtumor nach einer Operation
Unmöglichkeit einer chirurgischen Behandlung
Die stereotaktische Strahlentherapie weist bei spinalen Schwannomen eine hohe lokale Kontrollrate (über 90 %) und ein geringes Komplikationsrisiko auf.
Bei vestibulären Schwannomen:
SRS als Option bei kleinen bis mittelgroßen vestibulären Schwannomen, insbesondere bei Patienten, bei denen das Hauptziel darin besteht, die Funktion des Gesichts- und Hörnervs bei minimalem Eingriff zu erhalten
Wachstumskontrolle > 90 % bei einer 10–15-jährigen Überwachung nach einer Einzeldosis von 11–13 Gy
Das Risiko einer späteren Verschlechterung des Hörvermögens bleibt bestehen, ist jedoch in der Regel geringer als nach einem mikrochirurgischen Eingriff in den entsprechenden Kohorten.
Strahlentherapie bei NF2
Sie wird aufgrund des erhöhten Risikos für sekundäre Neoplasien (MPNST, Sarkome, Meningeome) bei Patienten mit Tumorsyndromen mit größerer Vorsicht eingesetzt.
Bei NF2 werden chirurgische Eingriffe und/oder eine zielgerichtete Therapie (Bevacizumab) bevorzugt; eine Strahlentherapie kommt zum Einsatz, wenn eine chirurgische Behandlung nicht möglich oder zu gefährlich ist.
Medikamentöse Therapie
Für sporadische, isolierte Schwannome (peripher oder spinal) liegen keine überzeugenden Belege für den Nutzen einer systemischen Therapie vor. Die Standardtherapie besteht aus einer Operation bzw. Überwachung, bei Bedarf wird eine Strahlentherapie eingesetzt.
Das einzige Arzneimittel mit einer soliden Evidenzbasis und einem nachweisbaren klinischen Nutzen bei NF2-Schwannomatosen ist Bevacizumab bei progressiven, funktionell signifikanten vestibulären Schwannomen (und teilweise auch bei anderen NF2-Tumoren).
Bevacizumab
Anti-VEGF-monoklonale Antikörper sind die wichtigste nachgewiesene systemische Behandlungsoption bei progressiven NF2-assoziierten vestibulären Schwannomen, die das Gehör gefährden oder zu einer Kompression des Hirnstamms führen.
Eine systematische Metaanalyse von 8 Kohorten (161 Patienten) ergab bei etwa 41 % ein partielles Ansprechen hinsichtlich des Tumorvolumens, bei 47 % eine Stabilisierung und bei 7 % ein Fortschreiten der Erkrankung. Bei etwa 20 % der Patienten wurde eine Verbesserung des Hörvermögens, bei 69 % eine Stabilisierung (keine Verschlechterung des Hörvermögens) festgestellt.
Die Langzeitsicherheit und die langfristige Wirksamkeit werden weiterhin untersucht.
Sonstige zielgerichtete und experimentelle Ansätze
Beim Einsatz des mTOR-Hemmers Everolimus erwies sich die Wirksamkeit bei NF2-assoziierten vestibulären Schwannomen als begrenzt und unbeständig.
Tyrosinkinase-Hemmer (Lapatinib, EGFR/ErbB-Hemmer) zeigten in kleinen Studien bei Patienten mit NF2 vereinzelte partielle Ansprechraten.
Derzeit laufen Studien mit dem ALK-Inhibitor Brigatinib und HDAC-Inhibitoren (z. B. AR-42) bei NF2-assoziierten Tumoren; derzeit handelt es sich dabei um experimentelle Optionen im Rahmen klinischer Studien.
Prognose der Erkrankung
Sporadische gutartige Schwannome (peripher, spinal, vestibulär) rezidivieren bei angemessener Behandlung selten, haben praktisch keinen Einfluss auf die Gesamtüberlebensrate und ermöglichen es, den neurologischen Status bei den meisten Patienten zu erhalten oder zu verbessern.
Nach einer totalen Resektion von spinalen Schwannomen treten Rezidive bei weniger als 5 % der Patienten auf.
Bei der NF2-Schwannomatose wird die Prognose durch die Vielzahl der Tumoren (vestibuläre Schwannome, Meningeome, Ependymome), fortschreitende Innenohrschwerhörigkeit, Gleichgewichtsstörungen und Polyneuropathie bestimmt. Trotz verbesserter Diagnostik und Therapie bleibt die Lebenserwartung im Vergleich zur Allgemeinbevölkerung verringert.
Eine Malignisierung des klassischen Schwannoms ist äußerst selten. MPNST entsteht in der Regel aus plexiformen Neurofibromen bei NF1 oder nach einer Strahlentherapie und hat eine deutlich schlechtere Prognose (5-Jahres-Überlebensrate 34–60 %).
Aktive Überwachung („Wait-and-Scan“-Strategie) bei asymptomatischen Schwannomen:
Sie ist bei sporadischen kleinen vestibulären und peripheren/spinalen Schwannomen bei fehlender Kompression kritischer Strukturen und guter Patientencompliance begründet und weit verbreitet.
Sie erfordert ein klares Protokoll zur MRT-Überwachung (mindestens zwei frühzeitige Untersuchungen zur Bestimmung des Wachstumstyps, anschließend wird das Intervall individuell angepasst).
Sie sollte mit einer ausführlichen Aufklärung des Patienten über das Risiko des Wachstums und die mögliche Notwendigkeit eines späteren Eingriffs einhergehen.
Wachstumsgeschwindigkeit von Schwannomen
Das durchschnittliche Wachstum der meisten Schwannome (einschließlich vestibulärer Schwannome) ist gering (≈ 1 mm/Jahr im Durchmesser oder ein geringer Volumenzuwachs), es gibt jedoch einen seltenen Subtyp schnell wachsender Tumoren, der rechtzeitig erkannt werden muss.
Periphere und spinale Schwannome
Die meisten peripheren und spinalen Schwannome wachsen langsam: etwa 5–10 % des Volumens pro Jahr.
In der Arbeit von El Sayed L et al. betrug bei einer Serie von 47 peripheren Schwannomen (sporadische Schwannome und bei Schwannomatose) das relative jährliche Wachstum etwa 34 % pro Jahr, wobei ≈ 19 % der Tumoren nicht wuchsen oder sich verkleinerten, während bei ≈ 13 % ein „schnelles“ Wachstum (> 2 cm³/Jahr und > 35 %/Jahr) festgestellt wurde.
In der Arbeit von Lubelski D. et al. wurden in einer Serie von 227 Patienten zum natürlichen Verlauf von Schwannomen des Plexus cervicalis (Halsgeflecht) sowie peripheren und spinalen Tumoren folgende Subtypen unterschieden: „langsames“ Wachstum (~5–10 % des Volumens pro Jahr), „mäßiges“ und „schnelles“ (> 80 % pro Jahr), wobei sich der Wachstumstyp oft bereits anhand von zwei Kontrolluntersuchungen bestimmen lässt.
3D-Animation: Wachstum eines spinalen extraduralen Schwannoms
Vestibuläre Schwannome (VS)
Eine systematische Übersicht von Yoshimoto Y. zum natürlichen Verlauf ergab Folgendes:
Wachstumsrate (jeder positive Zuwachs) ≈ 46 % bei einer durchschnittlichen Beobachtungsdauer von 38 Monaten
Rückgang 8 %
Durchschnittliche Wachstumsrate am maximalen Durchmesser ≈ 1,2 mm/Jahr
Eine aktuelle Übersicht von Paldor I. et al. zum Wachstum von VS liefert ähnliche Zahlen: durchschnittliches Wachstum 0,99–1,11 mm/Jahr. Dabei beträgt die erwartete Wachstumsrate ≈ 3 mm/Jahr, wenn der Tumor bereits bei der ersten Kontroll-MRT ein Wachstum gezeigt hat, und Faktoren für ein schnelles Wachstum (> 4 mm/Jahr) sind Zysten, Blutungen und eine Hormontherapie.
Die Bandbreite der Wachstumsraten ist groß: von keinem Wachstum oder sogar einer Verkleinerung bis hin zu 6–17 mm/Jahr in einzelnen Serien.
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Wichtige praktische Hinweise für den Arzt
Jede schnelle Veränderung der Größe, der Art der Schmerzen oder des neurologischen Status bei einem Patienten mit bekanntem Schwannom (insbesondere bei NF1) erfordert den Ausschluss von MPNST (MRT mit DWI, PET-CT, Biopsie).
Bei einem vestibulären Schwannom bei jungen Patienten sollte eine NF2-bedingte Schwannomatose aktiv ausgeschlossen werden (MRT des gesamten ZNS, genetische Untersuchung).
Bei asymptomatischen Patienten mit kleinen Schwannomen und einem stabilen klinischen und radiologischen Befund ist eine aktive Überwachung mit regelmäßigen MRT-Untersuchungen unbedenklich.
FAQ
1. Was ist ein Schwannom und handelt es sich dabei um Krebs?
Ein Schwannom (oder Neurinom) ist ein gutartiger Tumor, der sich aus den Schwann-Zellen der Hüllen der peripheren oder Hirnnerven entwickelt. Es handelt sich nicht um Krebs, der Tumor ist von einer Kapsel umgeben, wächst langsam und expansiv und wird äußerst selten bösartig (entartet zu einer malignen Form).
2. Was sind die Ursachen für die Entstehung eines Neurinoms?
Die Ätiologie der Erkrankung hängt mit Mutationen des NF2-Gens auf Chromosom 22 zusammen, die zu einem Funktionsverlust des Merlin-Proteins und einer Störung der Zellteilungskontrolle führen. Diese Mutationen können spontan auftreten (sporadische Fälle) oder im Rahmen genetischer Syndrome vererbt werden, z. B. bei der NF2-Schwannomatose (früher als Neurofibromatose Typ 2 bezeichnet) oder anderen Formen der Schwannomatose.
3. Welche Symptome treten bei einem vestibulären Schwannom (Akustikusneurinom) auf?
Für einen Tumor des 8. Hirnnervs ist die klassische klinische Trias charakteristisch: einseitige Innenohrschwerhörigkeit, Tinnitus (ständiges Ohrgeräusch) und Gleichgewichtsstörungen.
4. Wie gefährlich ist ein Schwannom und wie hoch ist die Sterblichkeitsrate?
Die Prognose bei dieser Erkrankung ist günstig, da es sich um einen gutartigen Tumor handelt, der in den meisten Fällen langsam wächst; daher ist die direkt damit verbundene Sterblichkeitsrate äußerst gering.
5. Was ist ein Neurinom der Cauda equina und welche Symptome treten dabei auf?
Es handelt sich um ein Schwannom, das im lumbosakralen Bereich der Wirbelsäule lokalisiert ist, wo die Nervenwurzeln verlaufen. Zu den Symptomen gehören Rückenschmerzen, radikuläre Empfindungsstörungen in den Beinen, manchmal Muskelschwäche im Bein und Funktionsstörungen der Beckenorgane.
6. Ist ein Schwannom im CT- oder Ultraschallbild zu erkennen?
Das wichtigste Diagnoseverfahren ist MRT. Im Ultraschall sind nur oberflächliche Schwannome der peripheren Nerven der Extremitäten oder des Halses zu erkennen; für tief liegende Tumoren ist diese Methode nicht geeignet. Die Computertomographie (CT) stellt Weichteile zwar schlechter dar, ist jedoch wichtig für die Beurteilung von Knochenveränderungen: Erweiterung des Gehörgangs, der Zwischenwirbellöcher oder Zerstörung der Wirbel.
7. Was ist ein Morton-Neurom des Fußes?
Ein Morton-Neurom ist kein echter Tumor aus Schwann-Zellen, sondern eine lokale Verdickung (Fibrose) der Hülle des N. plantaris des Fußes aufgrund chronischer Kompression. Im Gegensatz zu spinalen oder kranialen Schwannomen äußert es sich durch brennende Schmerzen zwischen den Zehen beim Gehen und birgt kein onkologisches Risiko.
8. Was passiert mit Schwannomen nach der Operation?
Bei vollständiger chirurgischer Entfernung des Tumors treten Rezidive sehr selten auf, in weniger als 5 % der Fälle. Wenn ein Teil der Kapsel zur Erhaltung der Nervenfunktion belassen wurde, ist ein weiteres Wachstum des Restgewebes möglich, was bei weniger als 30 % der Patienten auftritt.
Quellenverzeichnis
1.
VOKA 3D Anatomy & Pathology – Complete Anatomy and Pathology 3D Atlas (VOKA 3D Anatomie und Pathologie – Vollständiger 3D-Atlas der normalen und pathologischen Anatomie) [Internet]. VOKA 3D Anatomie und Pathologie.
Verfügbar unter: https://catalog.voka.io/
2.
Goldbrunner R, et al. EANO guideline on the diagnosis and treatment of vestibular schwannoma. Neuro‑Oncology. 2019;21(3):e8–e23.
3.
CNS Guidelines Committee. Evidence‑based guidelines for the treatment of adults with vestibular schwannoma (update 2025). Congress of Neurological Surgeons.
4.
Sergi B, et al. Management of vestibular schwannoma: a systematic review with focus on radiotherapy vs surgery vs wait‑and‑scan. Journal of Personalized Medicine. 2022;12:1616.
5.
Pontillo V, et al. Hearing preservation surgery for vestibular schwannoma: systematic review. 2024.
6.
Laufer I, Bilsky M. Intradural nerve sheath tumors. UpToDate, 2023 update.
Evans DG, et al. NF2‑related schwannomatosis. UpToDate, 2022 update.
9.
Evans DG, Mostaccioli S, Pang D, et al. ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis. Eur J Hum Genet. 2022;30(7):812-817.
10.
Lu VM, Ravindran K, Graffeo CS, et al. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: a systematic review and meta-analysis of treatment outcomes. J Neurooncol. 2019;144(2):239-248.
11.
EANS Task Force. Surgical management of large vestibular schwannomas: systematic review and recommendations. Acta Neurochir. 2020.
12.
Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967-1977.
13.
Lubelski D, Pennington Z, Ochuba A, et al. Natural History of Brachial Plexus, Peripheral Nerve, and Spinal Schwannomas. Neurosurgery. 2022;91(6):883-891.
14.
El Sayed L, Masmejean EH, Parfait B, Kalamarides M, Biau D, Peyre M. Natural history of peripheral nerve schwannomas. Acta Neurochir (Wien). 2020;162(8):1883-1889.
15.
Yoshimoto Y. Systematic review of the natural history of vestibular schwannoma. J Neurosurg. 2005;103(1):59-63. doi:10.3171/jns.2005.103.1.0059
16.
Paldor I, Chen AS, Kaye AH. Growth rate of vestibular schwannoma. J Clin Neurosci. 2016;32:1-8. doi:10.1016/j.jocn.2016.05.003
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io
Cookie Consent
Wir verwenden Cookies, um Ihr Surferlebnis zu verbessern, den Datenverkehr auf der Website zu analysieren und Inhalte bereitzustellen. Bitte wählen Sie aus, ob Sie alle Cookies akzeptieren oder nicht wesentliche Tracking-Methoden ablehnen möchten.
Cookie-Einstellungen
Verwalten Sie Ihre Cookie-Einstellungen unten:
Essentielle Cookies ermöglichen grundlegende Funktionen und sind für die ordnungsgemäße Funktion der Website erforderlich.
Name
Beschreibung
Dauer
Geolocation-Konfiguration
Dieses Cookie wird verwendet, um die Zustimmungseinstellungen basierend auf dem Standort des Besuchers zu speichern.
30 Tage
Cookie-Einstellungen
Dieses Cookie wird verwendet, um die Cookie-Zustimmungseinstellungen des Benutzers zu speichern.
30 Tage
Google reCAPTCHA hilft, Websites vor Spam und Missbrauch zu schützen, indem Benutzerinteraktionen durch Herausforderungen verifiziert werden.
Name
Beschreibung
Dauer
_GRECAPTCHA
Google reCAPTCHA setzt ein notwendiges Cookie (_GRECAPTCHA), wenn es ausgeführt wird, um seine Risikoanalyse bereitzustellen.
179 Tage
Statistik-Cookies sammeln Informationen anonym. Diese Informationen helfen uns zu verstehen, wie Besucher unsere Website nutzen.
Google Analytics ist ein leistungsstarkes Tool, das den Website-Verkehr verfolgt und analysiert, um fundierte Marketingentscheidungen zu treffen.
Enthält benutzerdefinierte Informationen, die vom Webentwickler über die Methode _setCustomVar in Google Analytics festgelegt wurden. Dieses Cookie wird jedes Mal aktualisiert, wenn neue Daten an den Google Analytics-Server gesendet werden.
2 Jahre nach letzter Aktivität
__utmx
Wird verwendet, um festzustellen, ob ein Benutzer in einen A/B- oder multivariaten Test einbezogen ist.
18 Monate
_ga
ID, die zur Identifizierung von Benutzern verwendet wird.
2 Jahre
_gali
Wird von Google Analytics verwendet, um festzustellen, welche Links auf einer Seite angeklickt werden.
30 Sekunden
_ga_
ID, die zur Identifizierung von Benutzern verwendet wird.
2 Jahre
_gid
ID, die verwendet wird, um Benutzer 24 Stunden nach der letzten Aktivität zu identifizieren
24 Stunden
_gat
Verwendet zur Überwachung der Anzahl der Serveranfragen an Google Analytics bei Verwendung des Google Tag Managers
1 Minute
_gac_
Enthält Informationen zu den Marketingkampagnen des Benutzers. Diese werden mit Google AdWords / Google Ads geteilt, wenn die Google Ads- und Google Analytics-Konten miteinander verknüpft sind.
90 Tage
__utma
ID, die zur Identifizierung von Benutzern und Sitzungen verwendet wird.
2 Jahre nach letzter Aktivität
__utmt
Wird verwendet, um die Anzahl der Anfragen an den Google Analytics-Server zu überwachen.
10 Minuten
__utmb
Wird verwendet, um neue Sitzungen und Besuche zu unterscheiden. Dieses Cookie wird gesetzt, wenn die GA.js-JavaScript-Bibliothek geladen wird und kein bestehendes __utmb-Cookie vorhanden ist. Das Cookie wird jedes Mal aktualisiert, wenn Daten an den Google Analytics-Server gesendet werden.
30 Minuten nach letzter Aktivität
__utmc
Wird nur mit alten Urchin-Versionen von Google Analytics und nicht mit GA.js verwendet. Wurde verwendet, um zwischen neuen Sitzungen und Besuchen am Ende einer Sitzung zu unterscheiden.
Ende der Sitzung (Browser)
__utmz
Enthält Informationen über die Verkehrsquelle oder Kampagne, die den Benutzer auf die Website geleitet hat. Das Cookie wird gesetzt, wenn das GA.js-JavaScript geladen wird, und aktualisiert, wenn Daten an den Google Analytics-Server gesendet werden.
6 Monate nach letzter Aktivität
Clarity ist ein Webanalyse-Dienst, der den Website-Verkehr verfolgt und berichtet.
Verbindet mehrere Seitenaufrufe eines Nutzers zu einer einzigen Clarity-Sitzungsaufzeichnung.
1 Tag
_clck
Speichert einen eindeutigen Benutzeridentifikator, um das Besucherverhalten durch Heatmaps und Sitzungsaufzeichnungen zu analysieren.
1 Jahr
ANONCHK
Gibt an, ob MUID an ANID übertragen wird, ein Cookie, das für Werbung verwendet wird. Clarity verwendet ANID nicht, daher ist dies immer auf 0 gesetzt.
Sitzung
CLID
Identifiziert, wann Clarity diesen Nutzer zum ersten Mal auf einer Website gesehen hat, die Clarity verwendet.
12 Monate
_clsk
Verbindet mehrere Seitenaufrufe eines Nutzers zu einer einzigen Clarity-Sitzungsaufzeichnung.
12 Monate
_clck
Speichert die Clarity-Benutzer-ID und Präferenzen, die einzigartig für diese Website sind und der gleichen Benutzer-ID zugeordnet werden.