Schwannoma: eziologia, epidemiologia, classificazione, clinica, diagnostica, trattamento e prognosi
Gontsov A.Chirurgo neuro-oncologico, MD
19 minuti di lettura·Febbraio 26, 2026
Questo articolo è solo a scopo informativo
Il contenuto di questo sito web, inclusi testi, grafici e altri materiali, è fornito solo a scopo informativo. Non sono da intendersi come consigli o indicazioni. Per quanto riguarda la tua specifica condizione medica o il tuo trattamento, ti invitiamo a consultare il tuo operatore sanitario.
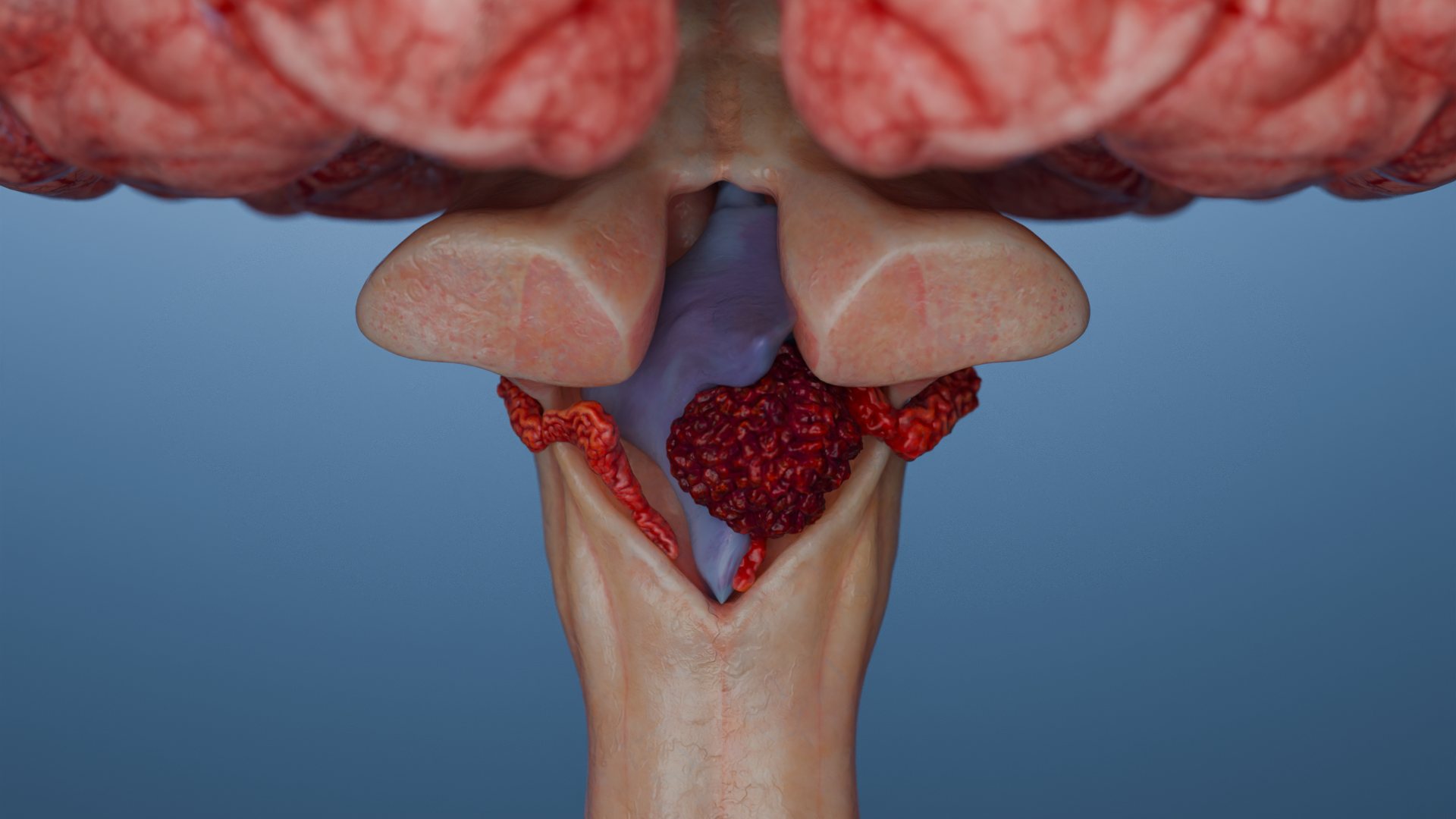
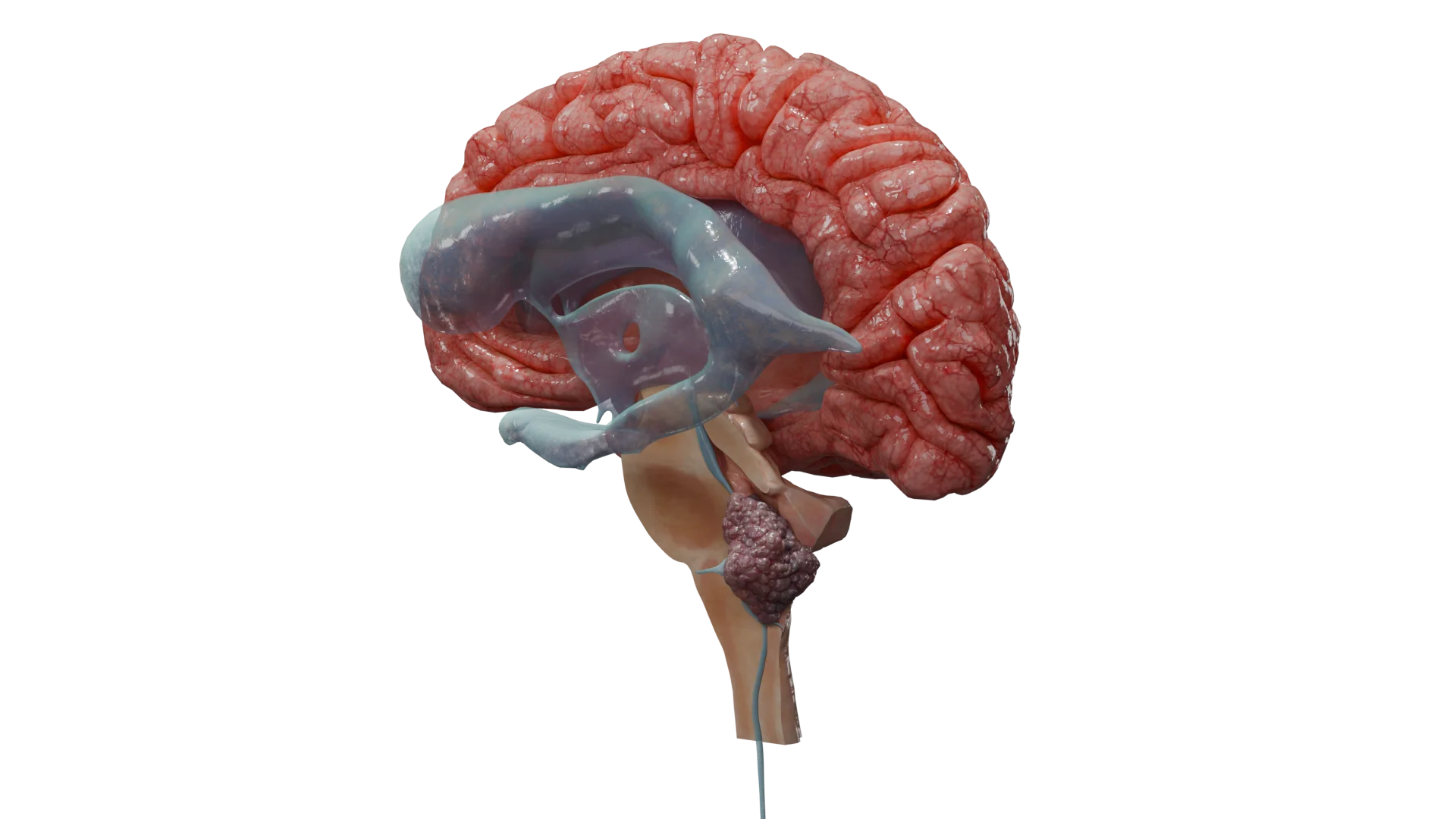
Lo schwannoma (detto anche neurinoma o neurilemoma) è un tumore benigno incapsulato che origina dalle cellule di Schwann periferiche e craniali, più frequentemente dell’VIII paio di nervi (schwannoma vestibolare – VS).
Nella maggior parte dei casi, lo schwannoma cresce lentamente, ha confini netti con il nervo (crescita espansiva) e raramente diventa maligno.
Gli schwannomi sporadici sono causati da mutazioni somatiche spontanee del gene NF2 sul cromosoma 22q che portano alla perdita della funzione della proteina merlina, con conseguente compromissione del controllo della proliferazione delle cellule di Schwann. La patogenesi della malattia corrisponde al modello dei “due colpi” (two-hit): una mutazione iniziale del gene NF2 in un allele e la successiva perdita di eterozigosi o un’altra inattivazione del secondo allele (LOH 22q, mutazioni puntiformi, ricombinazione mitotica, metilazione del promotore).
Nella schwannomatosi corellata a NF2 (NF2-related schwannomatosis, precedentemente nota come neurofibromatosi di tipo 2) l’allele mutante del gene NF2 è presente nella linea germinale. Gli eventi somatici aggiuntivi (detti anche “hit”) nel gene NF2 e in altri geni del cromosoma 22 causano lo sviluppo di schwannomi multipli (compresi quelli vestibolari bilaterali), meningiomi, ependimomi, ecc.
In altre forme di schwannomatosi (associate ai geni SMARCB1 e LZTR1), le mutazioni dei corrispondenti geni soppressori sul cromosoma 22q vengono ereditate. Per lo sviluppo del tumore è necessaria un’ulteriore inattivazione somatica del gene NF2.
Fattori di rischio dello schwannoma
Presenza di schwannomatosi del gene NF2, una sindrome ereditaria a trasmissione autosomica dominante che predispone agli schwannomi. Nei portatori della variante patogena del gene NF2, infatti, si formano inevitabilmente gli schwannomi vestibolari bilaterali e/o altri schwannomi multipli.
Presenza di schwannomatosi dei geni SMARCB1 o LZTR1, delle sindromi ereditarie caratterizzate da schwannomi multipli, prevalentemente non vestibolari, in assenza di schwannomi vestibolari bilaterali.
La neurofibromatosi di tipo 1 (NF1) non è associata agli schwannomi classici (nel gene NF1 predominano i neurofibromi e i MPNST) e gli schwannomi non sono considerati parte del gene NF1.
La precedente radioterapia è un fattore per lo sviluppo di MPNST, ma non di uno schwannoma tipico.
Epidemiologia
Gli schwannomi sono i tumori benigni più comuni dei nervi periferici.
Nella maggior parte dei casi, gli schwannomi sono tumori singoli; le lesioni multiple richiedono l’esclusione della schwannomatosi associata a NF2 e di altre forme di schwannomatosi.
Epidemiologia degli schwannomi spinali e periferici
Tra i tumori intradurali extramidollari del canale spinale, i nervi rivestiti da tumori costituiscono circa il 25%, di cui circa i 2/3 sono schwannomi, principalmente sporadici.
Solo il 2% circa di tutti gli schwannomi spinali riguarda pazienti con NF2, mentre gli altri sono casi sporadici o tumori singoli sintomatici.
Nella schwannomatosi NF2, i tumori spinali (prevalentemente gli schwannomi delle radici dorsali, ma anche i meningiomi e gli ependimomi) si riscontrano nel 90% dei pazienti nel corso della vita, rendendo gli schwannomi spinali uno dei segni epidemiologici chiave della malattia.
Gli schwannomi periferici (degli arti e dei plessi nervosi) sono rari nella popolazione generale. In una grande serie di centinaia di pazienti, rappresentano la forma principale di neoplasia benigna dei nervi periferici dei grandi tronchi.
Epidemiologia delle sindromi di predisposizione (nel contesto degli schwannomi)
Schwannomatosi NF2: negli studi condotti su grandi popolazioni, l’incidenza della malattia raggiunge un caso ogni 25.000 persone, con una quota significativa di casi rappresentata da forme a mosaico* (almeno la metà dei pazienti de novo).
Schwannomatosi dei geni SMARCB1 e LZTR1: i registri nazionali dei centri per lo studio della neurofibromatosi mostrano che si tratta di una malattia significativamente più rara rispetto alla schwannomatosi NF2, ma i pazienti affetti presentano un’elevata “carica” individuale di schwannomi. L’analisi dell’epidemiologia delle sindromi NF rivela che gli schwannomi costituiscono una minoranza rispetto alla schwannomatosi NF1 e NF2.
Spiegazione:
*Le forme a mosaico di schwannomatosi rappresentano le varianti della malattia in cui la variante patogena del gene (tipicamente NF2, SMARCB1 o LZTR1) è presente non in tutte le cellule dell’organismo, ma solo in alcuni cloni cellulari, originatisi dopo la fecondazione (dopo la formazione del zigote). In altre parole, la schwannomatosi è causata da mosaicismo somatico, ovvero la presenza di una mutazione in una parte dei tessuti, mentre l’altra parte rimane geneticamente normale.
Epidemiologia degli schwannomi vestibolari (VS/neurinoma acustico/schwannoma dell’angolo pontocerebellare)
Gli schwannomi vestibolari (VS) costituiscono l’80-90% dei tumori dell’angolo pontocerebellare negli adulti; circa il 4-6% dei casi è associato alla schwannomatosi NF2.
Circa il 90-95% dei pazienti con NF2 presenta schwannomi vestibolari bilaterali intorno ai 30 anni, mentre nella popolazione generale gli schwannomi vestibolari sono per lo più unilaterali e sporadici.
Nei bambini e nei giovani adulti con uno schwannoma vestibolare singolo, la percentuale di pazienti con mutazione germinale NF2 è significativamente più alta rispetto alle persone di età superiore ai 30 anni; pertanto, l’età rappresenta un importante indicatore epidemiologico della schwannomatosi NF2 latente.
Manifestazioni cliniche dello schwannoma
Schwannomi periferici (nervi e plessi periferici)
Sintomi:
nodo o tumore a crescita lenta lungo il nervo, indolore o doloroso;
parestesie;
ipoestesia locale o perdita di forza nella zona d’innervazione;
spesso si osserva il sintomo di Tinel (parestesie alla percussione).
Obiettivamente, si osserva una massa densa, elastica e mobile trasversalmente rispetto all’asse del nervo, ma fissa lungo il suo decorso. Il deficit neurologico più frequente è di tipo sensitivo, mentre quello motorio è meno comune.
Schwannomi spinali
Gli schwannomi spinali spesso hanno origine dalle radici dorsali (sensibili) e si sviluppano asintomaticamente per lungo tempo.
Spesso vengono rilevati occasionalmente durante un esame RM della colonna vertebrale o, meno frequentemente, durante un esame radiografico del torace.
Immagine di uno schwannoma spinale alla radiografia del torace
I sintomi tipici sono:
mal di schiena;
radiculopatie;
disturbi motori (debolezza muscolare al di sotto del livello di compressione del midollo spinale);
Schwannoma spinale a clessidra («hourglass/dumbbell») con compressione del midollo spinale
Schwannoma spinale a clessidra («hourglass/dumbbell») con compressione del midollo spinale
Schwannoma spinale a clessidra («hourglass/dumbbell») con compressione del midollo spinale
Schwannomatosi (NF2, SMARCB1 e LZTR1)
Gli schwannomi multipli sono spesso associati alla sindrome dolorosa, frequentemente caratterizzata da dolore neuropatico invalidante nella schwannomatosi SMARCB1/LZTR1. Nel caso della schwannomatosi NF2, invece, la malattia è spesso correlata alla presenza di meningiomi e tumori del midollo spinale.
Neurinoma vestibolare (acustico)
Triade classica: ipoacusia neurosensoriale unilaterale (o bilaterale nella schwannomatosi NF2), tinnito (rumore) e disturbi dell’equilibrio.
Nei casi di tumori di grandi dimensioni, si osservano mal di testa, atassia, sintomi da compressione del tronco cerebrale e idrocefalo, nonché lesione dei nervi facciale e trigemino.
Classificazione degli schwannomi
Per localizzazione
Schwannomi dei nervi periferici (arti e plessi);
Schwannomi spinali (intradurali/extradulari extramidollari). Frequenti (fino al 15% dei casi) sono gli schwannomi a clessidra (“hourglass/dumbbell”);
Schwannomi dei nervi craniali. Il più comune è lo schwannoma del nervo craniale VIII, noto anche come schwannoma vestibolare, schwannoma acustico o schwannoma dell’angolo pontocerebellare. Raramente si riscontrano gli schwannomi del V nervo (trigemino) e altri.
Animazione 3D – Schwannoma spinale a forma di manubrio («hourglass/dumbbell»)
Per causa (nel quadro delle caratteristiche clinico-genetiche)
Tipo
Segni principali
Schwannoma sporadico
Singolo, sviluppo benigno
Schwannomatosi NF2
Schwannomi vestibolari bilaterali + schwannomi multipli centrali (spinali) e periferici, meningiomi ed ependimomi
Schwannomatosi SMARCB1/LZTR1
Schwannomi multipli non addominali, senza schwannomi vestibolari (acustici) bilaterali, comunemente caratterizzati da una sindrome dolorosa neuropatica.
Per tipo istologico
Classico (convenzionale);
Altri tipi: cellulare, plessiforme, epitelioide e con alterazioni degenerative.
Tutti i tipi istologici di schwannomi menzionati sopra sono benigni.
Il tumore maligno delle guaine dei nervi periferici (MPNST) e il tumore maligno melanotico della guaina del nervo (malignant melanotic nerve sheath tumor – MMNST), precedentemente denominato schwannoma melanotico, rientrano nelle categorie distinte di tumori maligni della guaina del nervo e non sono classificati come schwannomi maligni secondo la terminologia WHO 2021.
Per gli schwannomi vestibolari esistono classificazioni specifiche, sia anatomiche (sistema di classificazione Koos, classificazione Hannover/Samii), basate sulla localizzazione e sulla dimensione del tumore, sia funzionali, basate sull’udito (Gardner-Robertson) e sulla funzione del nervo facciale (House–Brackmann).
Diagnosi del neurinoma
Quadro clinico ed esame
Valutazione della dinamica di sviluppo del tumore, del dolore e delle manifestazioni neurologiche; anamnesi familiare della schwannomatosi NF1/NF2.
Nel caso dello schwannoma vestibolare, si eseguono l’audiometria tonale, il test del linguaggio e, se necessario, dei test vestibolari.
Visualizzazione
La RM con contrasto è il metodo di scelta.
Neurinoma periferico/spinale: ipointenso o isointenso su T1, iperintenso su T2, accumula intensamente e spesso in modo omogeneo il mezzo di contrasto; spesso presenta una capsula evidente e una posizione eccentrica rispetto al nervo;
Neurinoma vestibolare: il tumore che accumula il contrasto nel condotto uditivo interno e si estende nell’angolo pontocerebellare (il cosiddetto “segno del cono gelato”).
La TC viene utilizzata per valutare le strutture ossee, come l’allargamento del canale osseo e del forame intervertebrale, e a volte si osserva la distruzione delle vertebre.
Verifica morfologica
Per i tumori superficiali e periferici si ricorre alla biopsia o alla rimozione completa del tumore con esame istologico: Antoni A/B, corpi di Verocay e positività diffusa per S100.
Nello schwannoma vestibolare tipico e in presenza del quadro classico alla RM nei pazienti immunocompetenti, la biopsia non è necessaria.
Test genetici
I test genetici non vengono utilizzati per verificare la presenza del tumore, ma per identificare o escludere la sindrome tumorale (schwannomatosi NF2, schwannomatosi SMARCB1/LZTR1), valutare il rischio familiare e programmare la sorveglianza.
Indicazioni:
schwannomi multipli;
giovane età;
schwannomi vestibolari bilaterali, associati a un altro tumore del SNC;
anamnesi familiare.
La differenziazione tra la schwannomatosi NF2 e la schwannomatosi SMARCB1/LZTR1 è fondamentale, in quanto determina la strategia di sorveglianza e il rischio di sviluppare altri tumori.
In caso di un singolo schwannoma sporadico in un adulto senza anamnesi familiare, il test di solito non è necessario.
Diagnosi differenziale
Neurofibroma (spesso in NF1): crescita intraneurale, fusiforme, non espansiva, senza una capsula evidente, presenza di assoni all’interno del tumore, rischio di malignizzazione (MPNST).
Tumori dei tessuti molli: lipoma, desmoide, ganglio, perineurioma, lesioni metastatiche e altre del nervo, paraganglioma, sarcoma.
Altri tumori intradurali extramidollari: meningioma, ependimoma mixopapillare ecc.
Nello schwannoma vestibolare: meningioma nell’angolo pontocerebellare, epidermoide, metastasi, raramente un tumore gliale.
Trattamento dello schwannoma
La strategia di trattamento dello schwannoma viene personalizzata considerando:
sintomi (dolore, deficit neurologico e compressione del midollo spinale);
dimensioni e dinamica di crescita;
stato dell’udito/funzione del nervo facciale (per gli schwannomi vestibolari);
contesto genetico (NF2/SMARCB1/LZTR1);
età e patologia concomitante.
Opzioni:
sorveglianza (wait‑and‑scan);
trattamento chirurgico;
radiochirurgia stereotassica (SRS) e radioterapia stereotassica (SRT);
terapia farmacologica (più spesso terapia mirata con bevacizumab): principalmente in caso di schwannomi di tipo NF2 o vestibolari in fase di progressione, non operabili.
Trattamento chirurgico
Schwannomi periferici e spinali
Indicazioni:
tumori sintomatici (dolore e manifestazioni neurologiche);
crescita rapida;
incertezza diagnostica;
sospetto di malignità.
Obiettivo: rimozione totale risparmiando la funzione; se necessario, è consentita la resezione subtotale per preservare la funzione nervosa.
Tecnica: microscopio, neuromonitoraggio intraoperatorio, identificazione e preservazione delle radici funzionali; nei tumori a forma di manubrio, accesso combinato.
Animazione 3D – Schwannoma spinale a forma di manubrio («hourglass/dumbbell»)
Risultati: dopo la resezione totale, le recidive sono rare (meno del 5%); nei casi di tumore residuo, la crescita continua si osserva nel 30% circa dei casi (più frequentemente nella schwannomatosi NF2), ma un secondo intervento chirurgico è necessario solo in una piccola percentuale di casi.
Schwannoma vestibolare
Le raccomandazioni moderne (EANO 2019, revisioni sistematiche) indicano una strategia che si basa su fattori dimensionali, sintomatologici e sullo stato dell’udito.
Per i tumori piccoli o medi con orecchio udente sono possibili tre strategie:
sorveglianza;
SRS;
microchirurgia con preservazione dell’udito e della funzione del nervo facciale.
Le revisioni sistematiche hanno dimostrato che la SRS garantisce un tasso di controllo a lungo termine del tumore superiore al 90% nei piccoli schwannomi vestibolari e risultati migliori nella preservazione dei nervi facciale e uditivo rispetto al trattamento chirurgico.
Per gli schwannomi vestibolari di grandi dimensioni (di solito superiore a 2,5-3 cm, con compressione del tronco encefalico) è raccomandata la resezione microchirurgica primaria con decompressione del tronco; gli accessi più comunemente utilizzati sono quelli retrosigmoideo e translabirintico.
La tendenza moderna del “cranial nerve sparing” consiste nella resezione subtotale seguita dalla SRS precoce sul residuo del tumore. Questo approccio ibrido, secondo i dati di serie multicentriche, garantisce un elevato controllo oncologico e i migliori risultati in termini di preservazione delle funzioni dei nervi VII e VIII.
Per gli schwannomi vestibolari associati a NF2:
Il trattamento chirurgico degli schwannomi vestibolari associati a NF2 è complicato dal loro carattere multifocale e dalla bassa vascolarizzazione, nonché dal rischio elevato di sordità bilaterale.
Dove possibile, si dà preferenza a interventi meno aggressivi, che preservano l’udito, o combinati, considerando il possibile effetto della terapia farmacologica (bevacizumab).
Radioterapia
Radiochirurgia stereotassica (SRS) e radioterapia stereotassica (SRT)
Per gli schwannomi periferici e spinali, le indicazioni sono le seguenti:
recidiva o tumore residuo post-operatorio;
il trattamento chirurgico non è possibile.
La radioterapia stereotassica ha dimostrato un elevato controllo locale (superiore al 90%) e un basso rischio di complicanze negli schwannomi spinali.
Per gli schwannomi vestibolari:
SRS: un’opzione per gli schwannomi vestibolari piccoli e medi, in particolare per i pazienti per i quali l’obiettivo principale è preservare la funzione dei nervi facciale e uditivo con un intervento minimo;
controllo della crescita superiore al 90% a 10-15 anni di follow-up con una singola dose di 11-13 Gy.
Il rischio di un peggioramento tardivo dell’udito persiste, ma è generalmente inferiore rispetto alla microchirurgia nelle coorti corrispondenti.
Radioterapia nella NF2
La radioterapia viene utilizzata con maggiore cautela a causa del rischio elevato di sviluppare neoplasie secondarie (MPNST, sarcomi e meningiomi) nei pazienti affetti da sindromi tumorali.
Nel caso della NF2, si dà la preferenza alla chirurgia e/o alla terapia mirata (bevacizumab), mentre la radioterapia viene utilizzata quando la chirurgia non è possibile o presenta dei rischi.
Terapia farmacologica
Per gli schwannomi sporadici e isolati (periferici o spinali) non ci sono dati convincenti sull’efficacia della terapia sistemica. Lo standard di trattamento prevede la combinazione di chirurgia e sorveglianza, con l’aggiunta di radioterapia quando necessario.
L’unico farmaco che presenta una solida base di evidenze scientifiche e un effettivo beneficio clinico nella schwannomatosi NF2 è il bevacizumab per schwannomi vestibolari progressivi, funzionalmente rilevanti (e parzialmente altri tumori NF2).
Bevacizumab
L’anticorpo monoclonale anti-VEGF rappresenta l’unica opzione sistemica approvata per schwannomi vestibolari NF2-progredienti, che presentano un rischio di perdita dell’udito o causano compressione del tronco.
Un’analisi sistematica di otto coorti (161 pazienti) ha evidenziato una risposta parziale nel volume del tumore del 41% circa, una stabilizzazione nel 47%, una progressione nel 7%, un miglioramento dell’udito nel 20% circa e una stabilizzazione (assenza di peggioramento uditivo) nel 69% dei pazienti.
Restrizioni della terapia con bevacizumab:
proteinuria;
ipertensione;
rischio di gravi complicanze vascolari (incluse rare emorragie intracraniche).
La sicurezza a lungo termine e l’efficacia duratura rimangono oggetto di studio.
Approcci alternativi mirati e sperimentali
L’inibitore mTOR everolimus: l’attività negli schwannomi vestibolari associati alla NF2 è risultata limitata e incostante.
Gli inibitori della tirosina chinasi (lapatinib e inibitori di EGFR/ErbB) hanno dimostrato risposte parziali isolate in piccoli studi su pazienti con NF2.
Sono in corso gli studi sull’uso dell’inibitore di ALK brigatinib e degli inibitori delle HDAC (come l’AR-42) nei tumori associati alla NF2; al momento, queste sono opzioni sperimentali nell’ambito di studi clinici.
Prognosi della malattia
Gli schwannomi benigni sporadici (periferici, spinali e vestibolari), se trattati adeguatamente, raramente recidivano, non influenzano significativamente la sopravvivenza complessiva e permettono di mantenere o migliorare lo stato neurologico nella maggior parte dei pazienti.
Dopo la resezione totale dello schwannoma spinale, le recidive si osservano in meno del 5% dei casi.
Nei casi di schwannomatosi associata a NF2, la prognosi è determinata dalla molteplicità dei tumori (schwannomi vestibolari, meningiomi ed ependimomi), dalla progressiva sordità neurosensoriale, dal disturbo dell’equilibrio e dalla polineuropatia. Nonostante i progressi significativi nella diagnosi e nella terapia, l’aspettativa di vita dei pazienti è più bassa rispetto a quella della popolazione generale.
La malignizzazione dello schwannoma classico è estremamente rara. Il MPNST origina principalmente a partire da neurofibromi plessiformi nella NF1 o a seguito di radioterapia e presenta una prognosi significativamente più sfavorevole (una sopravvivenza a cinque anni nel 34-60% dei casi).
La strategia di sorveglianza “wait and scan” per gli schwannomi asintomatici:
é giustificata e ampiamente accettata per piccoli schwannomi vestibolari e periferici/spinali sporadici in assenza di compressione di strutture critiche e con buona compliance del paziente;
richiede un protocollo di controllo con RM (almeno due esami iniziali per determinare il tipo di crescita; successivamente, l’intervallo tra gli esami viene personalizzato);
deve fornire al paziente informazioni dettagliate sui rischi di crescita e sull’eventuale necessità di interventi successivi.
Velocità di crescita dello schwannoma
La crescita media della maggior parte degli schwannomi (compresi quelli vestibolari) è lenta (circa 1 mm/anno in termini di diametro o un modesto aumento di volume), tuttavia esiste un raro sottotipo di tumore a crescita rapida che deve essere identificato tempestivamente.
Schwannomi periferici e spinali
Nella maggior parte dei casi, gli schwannomi periferici e spinali crescono lentamente, aumentando di circa il 5-10% del volume all’anno.
Uno studio di El Sayed L. et al. su una serie di 47 schwannomi periferici (sia sporadici che in caso di schwannomatosi) ha mostrato una crescita annuale relativa pari a circa il 34%. Circa il 19% dei tumori non cresceva o diminuiva, mentre il 13% circa mostrava una crescita “rapida” (superiore a 2 cm³ all’anno e al 35% all’anno).
Nello studio di Lubelski D. et al. su una serie di 227 pazienti, sono stati individuati i sottotipi di crescita degli schwannomi del plesso cervicale, dei tumori periferici e spinali in base al loro decorso naturale: crescita lenta (5-10% all’anno), moderata e rapida (superiore all’80% all’anno). Spesso, il tipo di crescita può essere determinato già nei primi due studi di controllo.
Animazione 3D – Crescita dello schwannoma spinale extradurale
Schwannomi vestibolari (VS)
Una revisione sistematica di Yoshimoto Y. sul decorso naturale ha mostrato:
la frequenza di crescita (qualsiasi aumento positivo) è di circa il 46% per un periodo medio di sorveglianza di 38 mesi;
la regressione dell’8%;
la velocità media di crescita in base al diametro massimo è di circa 1,2 mm/anno.
Una revisione moderna di Paldor I. et al. sulla crescita degli schwannomi vestibolari fornisce cifre simili: la crescita media è di 0,99-1,11 mm/anno. Inoltre, se il tumore ha già mostrato segni di crescita durante il primo controllo con RM, la velocità di crescita attesa è di circa 3 mm/anno. I fattori che possono causare una crescita rapida (superiore a 4 mm/anno) includono la presenza di cisti, emorragie e terapia ormonale.
L’intervallo di velocità è ampio: si va dall’assenza o addirittura dalla riduzione della crescita fino alla crescita di 6-17 mm/anno in singole serie.
Scopri altri contenuti scientificamente accurati sui nostri social network
Iscriviti e non perdere le ultime risorse
Questi sono gli aspetti fondamentali che il medico deve considerare
In caso di rapido aumento delle dimensioni o di cambiamento della natura del dolore o dello stato neurologico in un paziente con diagnosi già nota di schwannoma (soprattutto in caso di NF1), è necessario escludere la presenza di un MPNST (tramite RM con DWI, PET-TC e biopsia).
Nei pazienti giovani con schwannoma vestibolare, è imperativo escludere la schwannomatosi associata a NF2, utilizzando la risonanza magnetica di tutto il sistema nervoso centrale e test genetico.
Nei pazienti asintomatici che presentano piccoli schwannomi e un quadro clinico e radiologico stabile, una strategia sicura consiste nel monitoraggio con controlli periodici mediante RM.
FAQ
1. Cos’è lo schwannoma? È un cancro?
Lo schwannoma (detto anche neurinoma) è un tumore benigno che si sviluppa a partire dalle cellule di Schwann delle guaine dei nervi periferici o craniali. Non è un cancro, ha una capsula, si caratterizza per una crescita espansiva lenta e molto raramente si trasforma in forma maligna.
2. Quali sono le cause del neurinoma?
L’eziologia della malattia è legata alle mutazioni del gene NF2 sul cromosoma 22, che portano alla perdita della funzione della proteina merlina e all’alterazione del controllo della divisione cellulare. Queste mutazioni possono verificarsi spontaneamente (casi sporadici) o essere trasmesse ereditariamente nel contesto di sindromi genetiche, come nella schwannomatosi NF2 (precedentemente chiamata neurofibromatosi di tipo 2) o altre forme di schwannomatosi.
3. Quali sintomi provoca il neurinoma vestibolare (acustico)?
Un tumore della VIII coppia di nervi craniali è caratterizzato dalla classica triade clinica: ipoacusia neurosensoriale unilaterale, tinnito (rumore costante nell’orecchio) e disturbo dell’equilibrio.
4. Quanto è pericoloso lo schwannoma e quanto è alta la mortalità?
La prognosi è favorevole, poiché il tumore è benigno e nella maggior parte dei casi cresce lentamente, pertanto la mortalità direttamente correlata a esso è molto bassa.
5. Cos’è il neurinoma della cauda equina e quali sono i suoi sintomi?
È uno schwannoma localizzato nella regione lombosacrale, dove passano le radici nervose. I sintomi includono dolore alla schiena, disturbi radicolari della sensibilità nelle gambe, talvolta debolezza muscolare in una gamba e disfunzione degli organi pelvici.
6. Lo schwannoma è visibile alla TC o all’ecografia?
La tecnica diagnostica principale è la risonanza magnetica (RM). All’ecografia sono visualizzabili solo gli schwannomi superficiali dei nervi periferici delle estremità o del collo, mentre per i tumori profondi questa tecnica non è adeguata. La tomografia computerizzata (TC) visualizza i tessuti molli in modo meno efficace, ma è importante per valutare i cambiamenti ossei, come l’allargamento del dotto uditivo, dei fori intervertebrali o la distruzione delle vertebre.
7. Cos’è il neuroma di Morton del piede?
Il neuroma di Morton non è un tumore delle cellule di Schwann, ma un ispessimento locale (fibrosi) della guaina del nervo plantare del piede causato da una compressione cronica. A differenza degli schwannomi spinali o craniali, si manifesta con dolore bruciante tra le dita durante la deambulazione e non comporta rischi oncologici.
8. Cosa succede allo schwannoma dopo un intervento chirurgico?
In caso di rimozione chirurgica totale del tumore, le recidive sono molto rare, con una percentuale inferiore al 5% dei casi. Se invece una parte della capsula è stata lasciata per preservare la funzione del nervo, è possibile che il tessuto residuo continui a crescere, fenomeno che si osserva nel 30% circa dei pazienti.
Bibliografia
1.
VOKA 3D Anatomy & Pathology – Complete Anatomy and Pathology 3D Atlas (VOKA Anatomia e Patologia 3D – Atlante 3D completo di anatomia e patologia) [Internet]. VOKA 3D Anatomy & Pathology.
Disponibile su: https://catalog.voka.io/
2.
Goldbrunner R, et al. EANO guideline on the diagnosis and treatment of vestibular schwannoma. Neuro-Oncology. 2019;21(3):e8-e23 [Goldbrunner R, et al. EANO guideline on the diagnosis and treatment of vestibular schwannoma (Linee guida EANO sulla diagnosi e trattamento dello schwannoma vestibolare). Neuro‑Oncology. 2019;21(3):e8–e23].
3.
CNS Guidelines Committee. Evidence‑based guidelines for the treatment of adults with vestibular schwannoma (update 2025). Congresso dei Chirurghi Neurologici [CNS Guidelines Committee. Evidence‑based guidelines for the treatment of adults with vestibular schwannoma (update 2025) (Linee guida basate su evidenze per il trattamento di adulti con schwannoma vestibolare (aggiornamento 2025)). Congress of Neurological Surgeons].
4.
Sergi B, et al. Management of vestibular schwannoma: a systematic review with focus on radiotherapy vs surgery vs wait‑and‑scan. Journal of Personalized Medicine. 2022;12:1616 [Sergi B, et al. Management of vestibular schwannoma: a systematic review with focus on radiotherapy vs surgery vs wait‑and‑scan (Gestione dello schwannoma vestibolare: una revisione sistematica con focus su radioterapia vs chirurgia vs osservazione “wait-and-scan”). Journal of Personalized Medicine. 2022;12:1616].
5.
Pontillo V, et al. Hearing preservation surgery for vestibular schwannoma: systematic review [Pontillo V, et al. Hearing preservation surgery for vestibular schwannoma: systematic review (Chirurgia di preservazione dell’udito per schwannoma vestibolare: revisione sistematica). 2024]. 2024.
6.
Laufer I, Bilsky M. Intradural nerve sheath tumors. UpToDate, aggiornamento 2023 [Laufer I, Bilsky M. Intradural nerve sheath tumors (Tumori della guaina del nervo intradurale). UpToDate, 2023 update].
Evans DG, et al. NF2‑related schwannomatosis. UpToDate, aggiornamento 2022 [Evans DG, et al. NF2‑related schwannomatosis (Schwannomatosi associata a NF2). UpToDate, 2022 update].
9.
Evans DG, Mostaccioli S, Pang D, et al. ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis. Eur J Hum Genet. 2022;30(7):812-817 [Evans DG, Mostaccioli S, Pang D, et al. ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis (Linee guida cliniche ERN GENTURIS per la diagnosi, il trattamento, la gestione e la sorveglianza delle persone con schwannomatosi). Eur J Hum Genet. 2022;30(7):812-817].
10.
Lu VM, Ravindran K, Graffeo CS, et al. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: a systematic review and meta-analysis of treatment outcomes. J Neurooncol. 2019;144(2):239-248 [Lu VM, Ravindran K, Graffeo CS, et al. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: a systematic review and meta-analysis of treatment outcomes (Efficacia e sicurezza del bevacizumab per gli schwannomi vestibolari nella neurofibromatosi di tipo 2: una revisione sistematica e una meta-analisi degli esiti terapeutici). J Neurooncol. 2019;144(2):239-248].
11.
EANS Task Force. Surgical management of large vestibular schwannomas: systematic review and recommendations. Acta Neurochir. [EANS Task Force. Surgical management of large vestibular schwannomas: systematic review and recommendations (Gestione chirurgica dei grandi schwannomi vestibolari: revisione sistematica e raccomandazioni). Acta Neurochir. 2020]. 2020.
12.
Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24(9):1967-1977 [Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation (Criteri diagnostici e nomenclatura aggiornati per la neurofibromatosi di tipo 2 e la schwannomatosi: una raccomandazione di consenso internazionale). Genet Med. 2022;24(9):1967-1977].
13.
Lubelski D, Pennington Z, Ochuba A, et al. Natural History of Brachial Plexus, Peripheral Nerve, and Spinal Schwannomas. Neurosurgery. 2022;91(6):883-891. [Lubelski D, Pennington Z, Ochuba A, et al. Natural History of Brachial Plexus, Peripheral Nerve, and Spinal Schwannomas (Storia naturale degli schwannomi del plesso brachiale, spinali e dei nervi periferici). Neurosurgery. 2022;91(6):883-891].
14.
El Sayed L, Masmejean EH, Parfait B, Kalamarides M, Biau D, Peyre M. Natural history of peripheral nerve schwannomas. Acta Neurochir (Wien). 2020;162(8):1883-1889. [El Sayed L, Masmejean EH, Parfait B, Kalamarides M, Biau D, Peyre M. Natural history of peripheral nerve schwannomas (Storia naturale degli schwannomi dei nervi periferici). Acta Neurochir (Wien). 2020;162(8):1883-1889].
15.
Yoshimoto Y. Systematic review of the natural history of vestibular schwannoma. J Neurosurg. 2005;103(1):59-63. doi:10.3171/jns.2005.103.1.0059 [Yoshimoto Y. Systematic review of the natural history of vestibular schwannoma (Revisione sistematica della storia naturale dello schwannoma vestibolare). J Neurosurg. 2005;103(1):59-63. doi:10.3171/jns.2005.103.1.0059].
16.
Paldor I, Chen AS, Kaye AH. Growth rate of vestibular schwannoma. J Clin Neurosci. 2016;32:1-8. doi:10.1016/j.jocn.2016.05.003 [Paldor I, Chen AS, Kaye AH. Growth rate of vestibular schwannoma (Tasso di crescita dello schwannoma vestibolare). J Clin Neurosci. 2016;32:1-8. doi:10.1016/j.jocn.2016.05.003].
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Grazie!
Il tuo messaggio è stato inviato! I nostri esperti ti contatteranno a breve. Se hai altre domande, contattaci all'indirizzo info@voka.io.
Cookie Consent
Utilizziamo i cookie per migliorare la tua esperienza di navigazione, analizzare il traffico del sito e fornire contenuti. Ti preghiamo di scegliere se accettare tutti i cookie o se desideri rifiutare il tracciamento non essenziale.
Preferenze Cookie
Gestisci le tue preferenze sui cookie di seguito:
I cookie essenziali abilitano funzioni di base e sono necessari per il corretto funzionamento del sito web.
Nome
Descrizione
Durata
Configurazione Geolocalizzazione
Questo cookie viene utilizzato per memorizzare le impostazioni di consenso in base alla posizione del visitatore.
30 giorni
Preferenze Cookie
Questo cookie viene utilizzato per memorizzare le preferenze di consenso ai cookie dell'utente.
30 giorni
Google reCAPTCHA aiuta a proteggere i siti web dallo spam e dagli abusi verificando le interazioni degli utenti attraverso sfide.
Nome
Descrizione
Durata
_GRECAPTCHA
Google reCAPTCHA imposta un cookie necessario (_GRECAPTCHA) quando viene eseguito per fornire la propria analisi del rischio.
179 giorni
I cookie statistici raccolgono informazioni in modo anonimo. Queste informazioni ci aiutano a comprendere come i visitatori utilizzano il nostro sito web.
Google Analytics è uno strumento potente che traccia e analizza il traffico del sito web per decisioni di marketing informate.
Contiene informazioni personalizzate impostate dallo sviluppatore web tramite il metodo _setCustomVar in Google Analytics. Questo cookie viene aggiornato ogni volta che nuovi dati vengono inviati al server di Google Analytics.
2 anni dopo l'ultima attività
__utmx
Utilizzato per determinare se un utente è incluso in un test A/B o multivariato.
18 mesi
_ga
ID utilizzato per identificare gli utenti.
2 anni
_gali
Utilizzato da Google Analytics per determinare quali link su una pagina vengono cliccati.
30 secondi
_ga_
ID utilizzato per identificare gli utenti.
2 anni
_gid
ID utilizzato per identificare gli utenti per 24 ore dopo l'ultima attività.
24 ore
_gat
Utilizzato per monitorare il numero di richieste al server di Google Analytics quando si utilizza Google Tag Manager.
1 minuto
_gac_
Contiene informazioni relative alle campagne di marketing dell'utente. Queste vengono condivise con Google AdWords / Google Ads quando gli account Google Ads e Google Analytics sono collegati.
90 giorni
__utma
ID utilizzato per identificare utenti e sessioni
2 anni dopo l'ultima attività
__utmt
Utilizzato per monitorare il numero di richieste al server di Google Analytics
10 minuti
__utmb
Utilizzato per distinguere nuove sessioni e visite. Questo cookie viene impostato quando la libreria javascript GA.js viene caricata e non esiste un cookie __utmb esistente. Il cookie viene aggiornato ogni volta che i dati vengono inviati al server di Google Analytics.
30 minuti dopo l'ultima attività
__utmc
Utilizzato solo con le vecchie versioni Urchin di Google Analytics e non con GA.js. Veniva utilizzato per distinguere tra nuove sessioni e visite alla fine di una sessione.
Fine della sessione (browser)
__utmz
Contiene informazioni sulla fonte di traffico o sulla campagna che ha diretto l'utente al sito web. Il cookie viene impostato quando il javascript GA.js viene caricato e aggiornato quando i dati vengono inviati al server di Google Analytics.
6 mesi dopo l'ultima attività
Clarity è un servizio di analisi web che traccia e riporta il traffico del sito web.