Chemodectoma (paraganglioma): Etiologia, patogénese, classificação, diagnóstico, métodos de tratamento
Kizyukevich O.Cirurgião cardiovascular, MD
10 min ler·Maio 07, 2025
Este artigo é apenas para fins informativos
O conteúdo deste sítio Web, incluindo texto, gráficos e outros materiais, é fornecido apenas para fins informativos. Não se destina a servir de conselho ou orientação. Relativamente ao teu estado de saúde ou tratamento específico, consulta o teu profissional de saúde.
Paraganglioma do corpo carotídeo (quimiodectoma) é um tumor raro, predominantemente benigno, que origina-se de células quimiorreceptoras do corpo carotídeo localizado na junção da artéria carótida comum.
Aproximadamente 1-2 casos por 100.000 habitantes.
As mulheres são mais frequentemente afectadas do que os homens.
A idade média do diagnóstico é de cerca de 45 anos.
A prevalência é elevada em países da América Latina, especialmente no México, onde as mulheres representam até 90% dos casos.
Os habitantes de zonas de altitude elevada têm um risco acrescido de desenvolver quimiodectomas devido à hipóxia crónica.
Etiologia
Mutações genéticas — as mutações nos genes que codificam subunidades do complexo succinato desidrogenase (SDH), envolvido na cadeia respiratória mitocondrial e metabolismo celular, desempenham um papel importante. Principais genes:
SDHD — frequentemente associada a paragangliomas múltiplos na cabeça e pescoço; transmitida por via paterna;
SDHB — associada a formas mais agressivas e risco aumentado de evolução maligna;
SDHC, SDHA, SDHAF2 — menos frequentemente envolvidas, mas também podem contribuir para o desenvolvimento de tumores.
Síndromes, associadas a quimiodectomas:
síndrome de Von Hippel-Lindau (VHL);
neoplasia endócrina múltipla tipo 2 (MEN2);
neurofibromatose tipo 1 (NF1).
Hipóxia crônica — viver em altitudes superiores a 2000 m, bem como condições como doença pulmonar obstrutiva crônica e defeitos cardíacos congênitos, pode estimular a hiperplasia do corpo carotídeo.
História familiar — cerca de 10% dos casos são familiares.
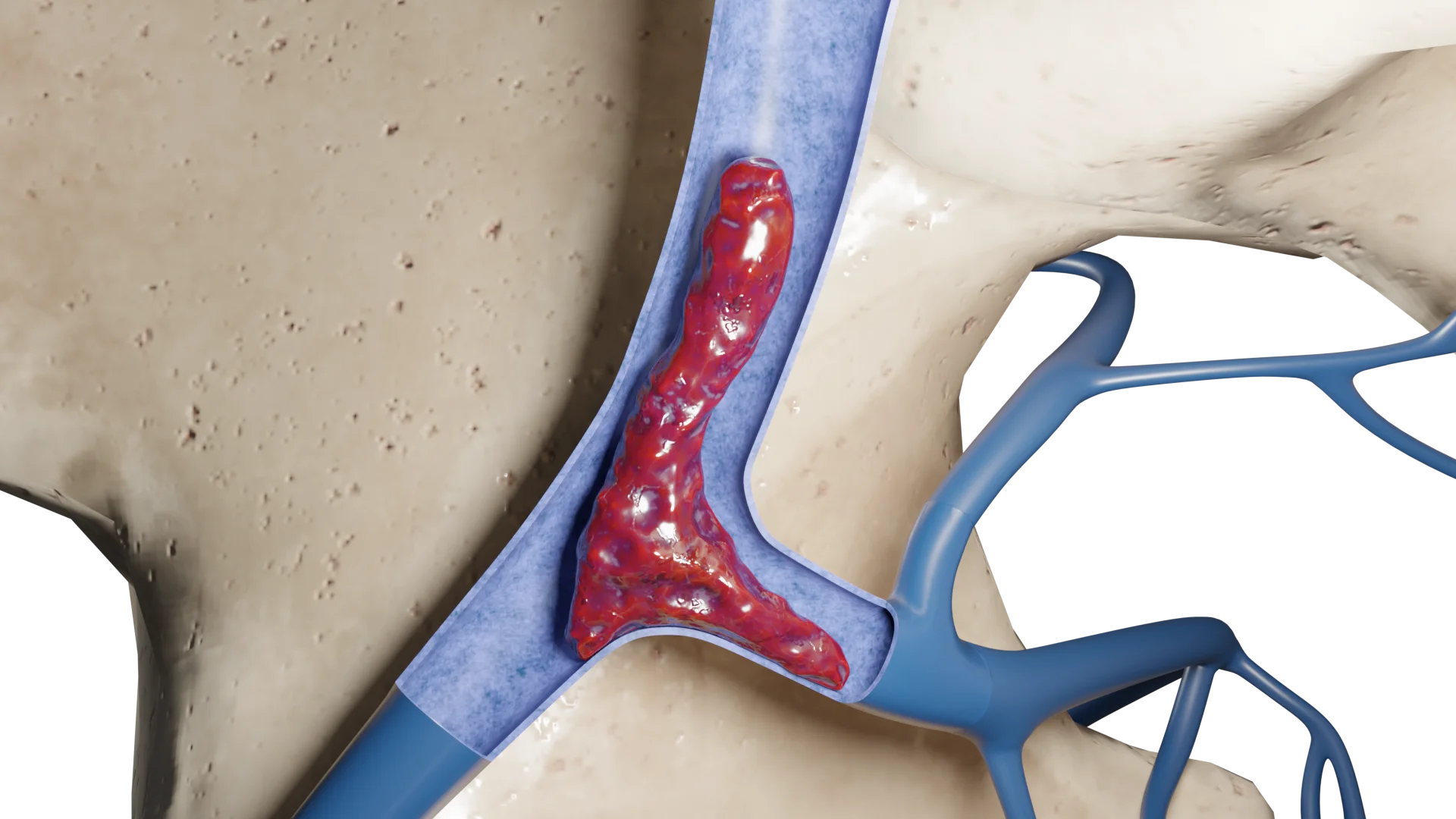
Animação 3D – Tipo I de quimiodectoma
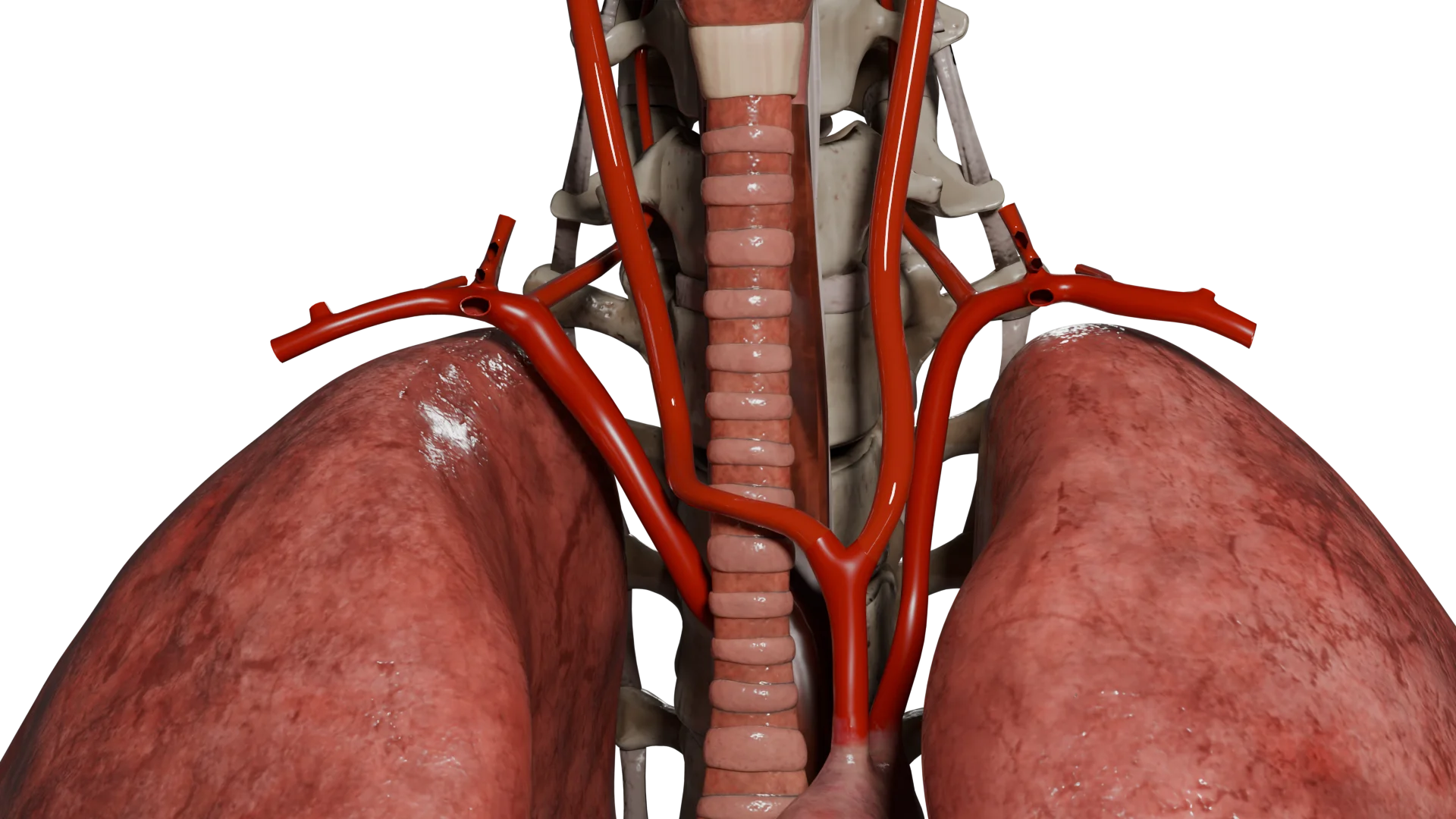
Animação 3D – Tipo III de quimiodectoma
Patogênese
Mutações hereditárias (SDHx)
Mutações SDH (enzima-chave do complexo II da cadeia respiratória mitocondrial e do ciclo de Krebs) → acumulação de succinato (atua como oncometabolito“oncometabolito”)} → inativação das prolil hidroxilases (PHDs) — enzimas que controlam a degradação do HIF-1α (fator induzido pela hipóxia) → acumulação de HIF-1α → pseudo-hipóxia → ativação do VEGF (angiogênese) / GLUT1 (glicólise) / PDGF (proliferação celular) → transformação neoplásica de células quimiorreceptoras e formação de tumor.
Hipóxia crónica
Hipóxia (altitude, DPOC) → diminuição da pO₂ → ativação dos receptores carótidos → hiperplasia celular → acumulação de HIF-1α → angiogênese e neoplasia.
Como resultado, o tumor aumenta gradualmente de tamanho. À medida que cresce, o tumor pode apresentar caráter invasivo local, envolvendo ou comprimindo estruturas anatômicas próximas, como artérias carótidas interna e externa, nervo vago, hipoglosso e glossofaríngeo. Isso pode causar sintomas neurológicos e vasculares associados.
Classificação do paraganglioma
William Shamblin propôs dividir paragangliomas do corpo carotídeo em três tipos (classificação de Shamblin):
Tipo I: Tumores limitados até 3,5 cm, vagamente associados às paredes arteriais;
Tipo II: 3,5–5 cm, cobrem parcialmente artérias carótidas, com fusão mais densa com as paredes arteriais;
Tipo III: Mais de 5 cm, envolve artérias carótidas e/ou estruturas vasculares e nervosas adjacentes, com fusão acentuada com estas estruturas.
Além disso, o quimiodectoma pode ser classificado pela etiologia: esporádico (até 85%), familiar (10–15%), hiperplásico (1–5%).
Quimiodectoma Tipo I
Quimiodectoma Tipo III
Manifestações clínicas
Durante muito tempo, pode não haver sintomas. À medida que o tumor cresce, podem surgir queixas:
tumor palpável — massa de crescimento lento, indolor, pulsante na superfície lateral do pescoço (na área do esternocleidomastóideo);
sintomas neurológicos — rouquidão da voz, disfagia, dormência da língua devido à compressão dos nervos cranianos (IX–XII);
queixas relacionadas à liberação de catecolaminas — palpitaciones, crises de hipertensão arterial, sudorese, dor de cabeça, tremores;
manifestações raras — tontura, desmaios durante a compressão do seio carotídeo.
Diagnóstico do paraganglioma
Exame físico: Tumor denso e pulsante no pescoço, que se desloca horizontalmente, mas não verticalmente.
Ecografia com Doppler: Tumor hipervascular na bifurcação da carótida.
RM com contraste: Visão característica de “sal e pimenta” em imagens ponderadas em T1.
TC com contraste: Determinação do grau de invasão vascular, avaliação da classificação de Shamblin. Construção de um modelo 3D para o planeamento da cirurgia.
Angiografia: Identificação do “sinal da lira” — divergência das artérias carótidas interna e externa. Permite avaliar a necessidade/possibilidade de embolização como etapa antes da cirurgia.
PET-CT com68Ga-DOTATATE: Se houver suspeita de lesões múltiplas ou metastáticas.
Exames laboratoriais: Determinação de metanefrinas e normetanefrinas em plasma e urina na suspeita de um tumor secretor.
Teste genético para mutações nos genes SDH se houver história familiar ou tumores múltiplos.
Encontra mais conteúdos cientificamente exactos nas nossas redes sociais
Subscreve e não percas os recursos mais recentes
Tratamento
O tratamento do quimiodectoma carotídeo depende do tamanho do tumor, dos sintomas clínicos, do risco de complicações, da atividade funcional, do perfil genético e do grau de Shamblin. As principais modalidades de tratamento são a excisão cirúrgica, a embolização pré-operatória e, em casos selecionados, a radioterapia ou a quimioterapia.
Embolização pré-operatória
A embolização pré-operatória é realizada para reduzir a vascularização do tumor, diminuir a perda sanguínea durante a cirurgia e facilitar a remoção do tumor.
Indicações:
tumores de Shamblin de grau II-III;
diâmetro do tumor > 3 cm;
presença de fluxo sanguíneo arterial significativo (de acordo com a TC/angiografia);
ressecção planeada com reconstrução vascular.
Contra-indicações:
tumores de Shamblin I;
não tem componente arterial (pouca vascularização);
anastomoses com a circulação cerebral – risco de embolização cerebral.
É realizada angiografia seletiva da artéria carótida externa. A introdução de agentes embolizantes é realizada através de microcateter: partículas de álcool polivinílico (PVA), microesferas, raramente cola ou espirais. A angiografia de controlo confirma a redução do fluxo sanguíneo. A cirurgia é efectuada no prazo de 24-48 horas após a embolização, enquanto o efeito persistir.
Tratamento cirúrgico
Indicações:
tumores sintomáticos (dor, disfagia, perturbação da fala);
crescimento rápido do tumor;
tumores > 2,5–3 cm (mesmo assintomáticos);
atividade confirmada por PET ou bioquímica;
mutação SDHB confirmada (risco de malignidade);
idade < 60 anos.
Tipos de tratamento cirúrgico:
Ressecção extracapsular do tumor. Padrão para Shamblin I-II, preservação vascular, risco mínimo.
Ressecção da artéria carótida com reconstrução. Mais frequentemente com Shamblin III. Após a ressecção da artéria carótida, é realizada uma prótese (Dacron, PTFE).
Neuromonitorização e microcirurgia. Utiliza-se quando está próximo dos nervos cranianos (IX-XII).
Possíveis complicações:
hemorragia (especialmente em caso de embolização inadequada);
danos nos nervos cranianos (IX–XII) – até 30% em tumores grandes;
acidente vascular cerebral (quando o fluxo sanguíneo colateral é prejudicado);
recidiva (rara, com ressecção incompleta).
Quimioterapia
Não se trata de um tratamento padrão para o quimiodectoma carotídeo, uma vez que a maioria dos tumores é de crescimento lento e de natureza benigna. No entanto, em casos raros de evolução maligna (progressão rápida, lesões metastáticas) ou em formas metastáticas inoperáveis, pode ser utilizada terapia sistémica.
Terapia com radionuclídeos ¹⁷⁷Lu-DOTATATE (PRRT)
Prescrito na presença de expressão de receptores de somatostatina por PET com ⁶⁸⁸Ga-DOTATATE.
FAQ
1. A chemodectomia carotídea é um tumor maligno?
Na maioria dos casos – não. A chemodectomia carotídea é considerada um tumor benigno de crescimento lento. No entanto, na presença de mutações do gene SDHB, pode haver potencial maligno com metástase.
2. É possível apenas observar a chemodectomia carotídea sem removê-la?
Sim, em alguns casos é possível a vigilância ativa, especialmente se o tumor for pequeno ( < 2,5 cm), assintomático, o paciente for idoso ou houver contraindicações à cirurgia. No entanto, se houver sinais de crescimento ou compressão das estruturas do pescoço, o tratamento é necessário.
3. A cirurgia para remoção da chemodectomia carotídea é perigosa?
Os riscos dependem do tamanho e da localização do tumor. Em casos de massas grandes, complicações podem ocorrer, incluindo danos aos nervos cranianos, sangramentos e eventos isquêmicos. Para minimizar os riscos, uma embolização pré-operatória é frequentemente realizada.
4. É necessário realizar exames em outros membros da família se eu tiver sido diagnosticado com chemodectomia?
Se você tiver uma mutação hereditária identificada (por exemplo, SDHD, SDHB), aconselha-se o aconselhamento genético e a investigação nos parentes próximos, pois a doença pode ser familiar.
5. Como diferenciar a chemodectomia carotídea de outros tumores do pescoço?
A chemodectomia carotídea geralmente está localizada na área de bifurcação da artéria carótida comum, pulsa, desloca-se horizontalmente, mas não verticalmente, e mostra um “sinal de lira” característico em TC/MRI. O diagnóstico é refinado através de imagiologia e angiografia.
6. A chemodectomia pode causar dor ou desconforto?
Na maioria dos casos – não, especialmente nos estágios iniciais. No entanto, à medida que o tumor cresce, pode pressionar nervos e vasos, levando a dor, rouquidão, dificuldade de engolir ou tontura.
7. Existe risco de recorrência após a remoção do tumor?
Com a remoção completa do tumor, o risco de recorrência é baixo (menos de 5%). No entanto, com ressecção incompleta, forma hereditária ou presença de mutação SDHB, pode haver recorrência ou desenvolvimento de novos focos. Nesses casos, a observação a longo prazo é importante.
Referências
1.
VOKA 3D Anatomy & Pathology – Complete Anatomy and Pathology 3D Atlas (VOKA 3D Anatomia e Patologia – Atlas 3D completo de anatomia e patologia) [Internet]. VOKA 3D Anatomy & Pathology [VOKA 3D Anatomia & Patologia].
Disponível em: https://catalog.voka.io/
2.
Luna-Ortiz K, Reynoso-Noverón N, Herrera-Ponzanelli C, Favila-Lira S, Luna-Peteuil Z, Herrera-Gomez A, et al. Diferenças sexuais de acordo com a apresentação étnica em tumores do corpo carotídeo: uma revisão sistemática da literatura. Int J Otorhinolaryngol Head Neck Surg. 2022 Jun;8(6):527-531. doi: 10.18203/issn.2454-5929.ijohns20221393.
3.
Butt N, Baek WK, Lachkar S, Iwanaga J, Mian A, Blaak C, et al. O corpo carotídeo e tumores associados: revisão atualizada com relevância clínica/cirúrgica. Br J Neurosurg. 2019 Out;33(5):500-503. doi: 10.1080/02688697.2019.1617404.
4.
Darouassi Y, Alaoui M, Mliha Touati M, Al Maghraoui O, En-Nouali A, Bouaity B, Ammar H. Tumores do corpo carotídeo: uma série de casos e revisão da literatura. Ann Vasc Surg. 2017 Aug;43:265-271. doi: 10.1016/j.avsg.2017.03.167.
5.
Gonzalez-Urquijo M, Castro-Varela A, Barrios-Ruiz A, Hinojosa-Gonzalez DE, Salas AKG, Morales EA, et al. Tendências atuais em tumores do corpo carotídeo: revisão abrangente. Head Neck. 2022 Out;44(10):2316-2332. doi: 10.1002/hed.27147.
6.
Ozawa H. Gestão atual de tumores do corpo carotídeo. Auris Nasus Larynx. 2024 Jun;51(3):501-506. doi: 10.1016/j.anl.2024.01.007.
7.
Piazza C, Lancini D, Tomasoni M, Zafereo M, Poorten VV, Hanna E, et al. Tumores malignos do corpo carotídeo: o que sabemos, o que fazemos e o que precisamos atingir. Uma revisão sistemática da literatura. Head Neck. 2024 Mar;46(3):672-687. doi: 10.1002/hed.27624.
São Petersburgo FL 33702, 7901 4th St N STE 300, EUA
Obrigado!
A tua mensagem foi enviada! Os nossos especialistas entrarão em contacto contigo em breve. Se tiveres mais perguntas, contacta-nos através de info@voka.io
Cookie Consent
Utilizamos cookies para melhorar a sua experiência de navegação, analisar o tráfego do site e fornecer conteúdo. Por favor, escolha se aceita todos os cookies ou se prefere recusar o rastreamento não essencial.
Preferências de Cookies
Gira as suas preferências de cookies abaixo:
Os cookies essenciais permitem funcionalidades básicas e são necessários para o funcionamento correto do site.
Nome
Descrição
Duração
Definições de geolocalização
Este cookie é utilizado para armazenar as configurações de consentimento com base na localização do visitante.
30 dias
Preferências de Cookies
Este cookie é utilizado para armazenar as preferências de consentimento do utilizador em relação aos cookies.
30 dias
O Google reCAPTCHA ajuda a proteger sites contra spam e abusos, verificando as interações dos usuários através de verificações de segurança.
Nome
Descrição
Duração
_GRECAPTCHA
O Google reCAPTCHA define o cookie necessário (_GRECAPTCHA) quando é executado, para fornecer uma análise de risco.
179 dias
Os cookies estatísticos recolhem informações de forma anónima. Estas informações ajudam-nos a compreender como os visitantes utilizam o nosso site.
O Google Analytics é uma ferramenta poderosa que rastreia e analisa o tráfego do site para apoiar decisões de marketing fundamentadas.
Identificador utilizado para identificar utilizadores e sessões.
2 anos após a última atividade
__utmt
Utilizado para monitorizar o número de pedidos ao servidor do Google Analytics.
10 minutos
__utmb
Utilizado para distinguir sessões e visitas novas. Este cookie é definido quando a biblioteca GA.js é carregada e não existem cookies __utmb presentes. O cookie é atualizado sempre que dados são enviados para o servidor do Google Analytics.
30 minutos após a última atividade
__utmc
Utilizado apenas com versões antigas do Urchin do Google Analytics e não com GA.js. Era utilizado para distinguir entre sessões e visitas novas no final da sessão.
Fim da sessão (navegador)
__utmz
Contém informações sobre a origem do tráfego ou a campanha que direcionou o utilizador para o site. O cookie é definido quando o JavaScript GA.js é carregado e é atualizado ao enviar dados para o servidor do Google Analytics.
6 meses após a última atividade
__utmv
Contém informações personalizadas definidas pelo desenvolvedor através do método _setCustomVar no Google Analytics. Este cookie é atualizado sempre que novos dados são enviados para o servidor do Google Analytics.
2 anos após a última atividade
__utmx
Utilizado para determinar se o utilizador está incluído em um teste A/B ou em um teste multivariado.
18 meses
_ga
Identificador utilizado para distinguir utilizadores
2 anos
_gali
Utilizado pelo Google Analytics para identificar os links que são clicados na página
30 segundos
_ga_
Identificador utilizado para distinguir utilizadores
2 anos
_gid
Identificador utilizado para distinguir utilizadores durante 24 horas após a última atividade
24 horas
_gat
Utilizado para monitorizar o número de pedidos ao servidor do Google Analytics ao utilizar o Google Tag Manager.
1 minuto
_gac_
Contém informações relacionadas com as campanhas de marketing do utilizador. É partilhado com o Google Ads / Google Ads quando as contas do Google Ads e do Google Analytics estão ligadas.
90 dias
Clarity é um serviço de análise web que acompanha o tráfego do site e gera relatórios sobre o mesmo.
Vincula visualizações de várias páginas por um único utilizador numa única sessão do Clarity.
Um dia
_clck
Armazena um identificador único de utilizador para analisar o comportamento dos visitantes através de mapas de calor e gravações de sessões.
Um ano
ANONCHK
Indica se o MUID foi transferido para ANID, que é um cookie utilizado para publicidade. O Clarity não utiliza ANID, portanto, este é sempre definido como 0.
Sessão
CLID
Determina a primeira vez que o Clarity viu este utilizador em qualquer site que utiliza o Clarity.
12 meses
_clsk
Vincula visualizações de várias páginas por um único utilizador numa única sessão do Clarity.
12 meses
_clck
Mantém o identificador de utilizador Clarity e as preferências, únicas para esse site atribuídas ao mesmo identificador de utilizador.