Quimiodectoma (paraganglioma): Etiología, patogenia, clasificación, diagnóstico, métodos de tratamiento
Kizyukevich O.Cirujano cardiovascular, MD
10 min leer·mayo 07, 2025
Este artículo sólo tiene fines informativos
El contenido de este sitio web, incluidos textos, gráficos y otros materiales, se proporciona únicamente con fines informativos. No pretende ser un consejo u orientación. En relación con tu enfermedad o tratamiento específico, consulta a tu médico.
El paraganglioma del cuerpo carotídeo (quimiodectoma) es un tumor raro, principalmente benigno, que surge de las células quimiorreceptoras del cuerpo carotídeo ubicado en la bifurcación de la arteria carótida común.
Aproximadamente 1-2 casos por 100.000 habitantes.
Las mujeres se ven afectadas con más frecuencia que los hombres.
La edad media de diagnóstico es de unos 45 años.
La alta prevalencia se observa en los países de América Latina, especialmente en México, donde las mujeres constituyen hasta el 90% de los casos.
Los habitantes de zonas de gran altitud tienen un mayor riesgo de desarrollar quimiodectomas debido a la hipoxia crónica.
Etiología
Mutaciones genéticas — desempeñan un papel destacado las mutaciones en los genes que codifican las subunidades del complejo succinato deshidrogenasa (SDH), que participa en la cadena respiratoria mitocondrial y el metabolismo celular. Genes principales:
SDHD — más comúnmente asociado a paragangliomas múltiples de cabeza y cuello; de transmisión paterna;
SDHB — asociado a formas más agresivas y mayor riesgo de curso maligno;
SDHC, SDHA, SDHAF2 — están implicados con menor frecuencia, pero también pueden contribuir al desarrollo tumoral.
Síndromes, asociados con los quimiodectomas:
síndrome de Von Hippel-Lindau (VHL);
neoplasia endocrina múltiple tipo 2 (MEN2);
neurofibromatosis tipo 1 (NF1).
Hipoxia crónica — vivir a más de 2000 m sobre el nivel del mar, así como afecciones como la enfermedad pulmonar obstructiva crónica y las cardiopatías congénitas, pueden estimular la hiperplasia del cuerpo carotídeo.
Antecedentes familiares — alrededor del 10 % de los casos son familiares.
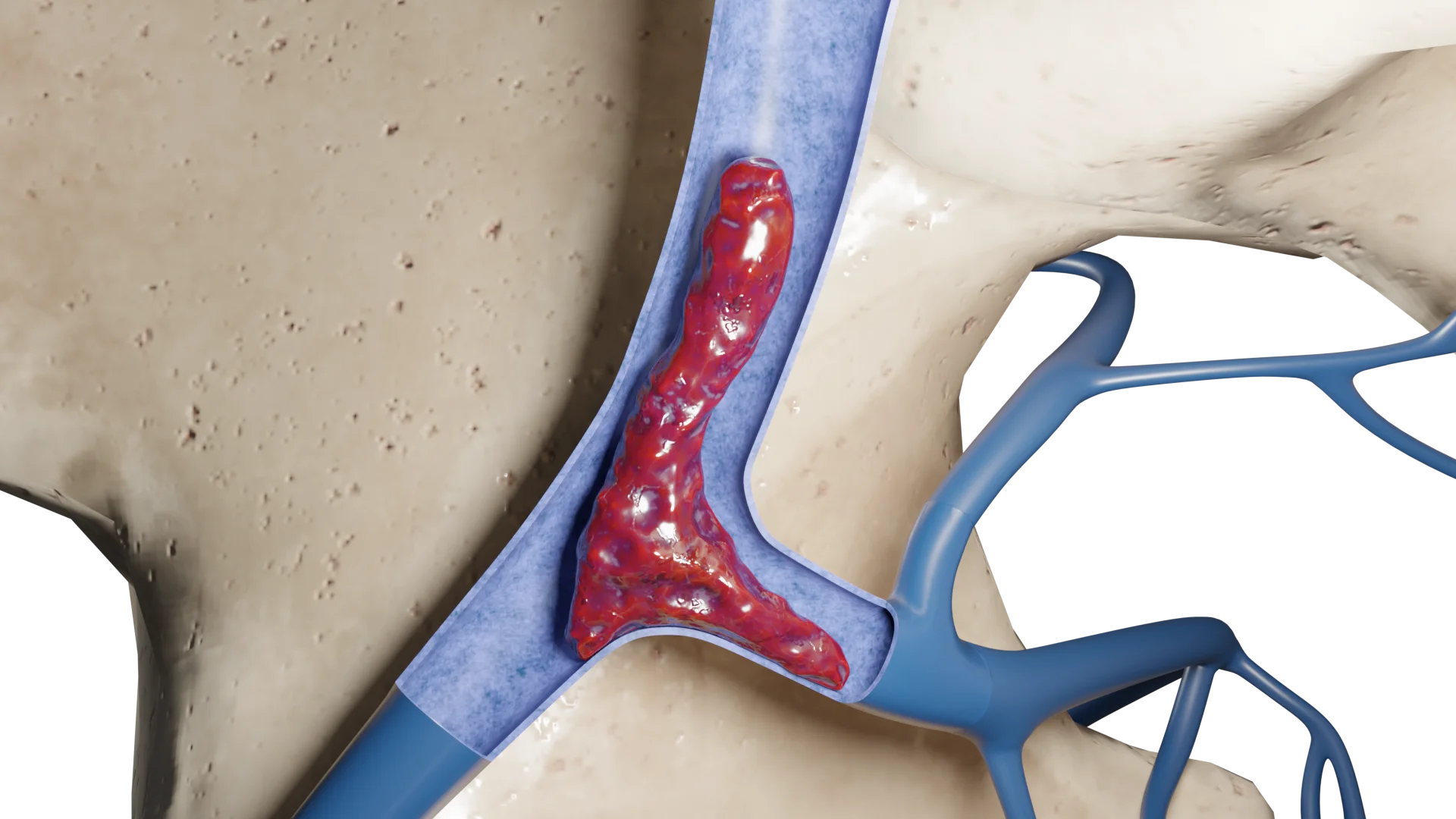
Animación 3D – quimiodectoma tipo I
Animación 3D – quimiodectoma tipo III
Patogénesis
Mutaciones hereditarias (SDHx)
Mutaciones SDH (enzima clave del complejo II de la cadena respiratoria mitocondrial y del ciclo de Krebs) → acumulación de succinato (actúa como oncometabolito) → inactivación de las prolil hidroxilasas (PHDs) — enzimas que controlan la degradación de HIF-1α (factor inducible por hipoxia) → acumulación de HIF-1α → pseudohipoxia → activación de VEGF (angiogénesis) / GLUT1 (glucólisis) / PDGF (proliferación celular) → transformación neoplásica de las células quimiorreceptoras y formación de tumores.
Hipoxia crónica
Hipoxia (altitud, EPOC) → Disminución de la pO₂ → Activación de los receptores carotídeos → Hiperplasia celular → Acumulación de HIF-1α → Angiogénesis y neoplasia.
Como resultado, el tumor aumenta gradualmente de tamaño. A medida que crece, puede mostrar un carácter localmente invasivo, hasta rodear o comprimir estructuras anatómicas cercanas, como las arterias carótidas interna y externa, los nervios vago, hipogloso y glosofaríngeo. Esto puede provocar síntomas neurológicos y vasculares asociados.
Clasificación del paraganglioma
William Shamblin propuso dividir los paragangliomas del cuerpo carotídeo en tres tipos (clasificación de Shamblin):
tipo I: tumores limitados de hasta 3,5 cm, escasamente asociados a las paredes arteriales;
tipo II: 3,5–5 cm, cubren parcialmente las arterias carótidas, tienen una fusión más estrecha con las paredes arteriales;
tipo III: más de 5 cm, cubren las arterias carótidas y/o los vasos cercanos, los nervios tienen una fusión marcadamente densa con estas estructuras.
Además, el quimiodectoma puede clasificarse por etiología: esporádica (hasta 85%), familiar (10–15%), hiperplásica (1–5%).
Quemodectoma Tipo I
Quemodectoma Tipo III
Manifestaciones clínicas
Puede no haber síntomas durante mucho tiempo. A medida que el tumor crece, pueden aparecer molestias:
tumor palpable — masa de crecimiento lento, indolora y pulsátil en la superficie lateral del cuello (en la zona del músculo esternocleidomastoideo);
síntomas neurológicos — ronquera de la voz, disfagia, adormecimiento de la lengua, causados por la compresión de los nervios craneales (IX–XII);
molestias relacionadas con la producción de catecolaminas — palpitaciones, episodios de hipertensión arterial, sudoración, dolor de cabeza, temblores;
manifestaciones poco frecuentes — mareos, desmayos al comprimir el seno carotídeo.
Diagnóstico del paraganglioma
Examen físico: tumor denso y pulsátil en el cuello, desplazable horizontalmente pero no verticalmente.
Ecografía con Doppler: tumor hipervascular en la zona de bifurcación de la arteria carótida.
RM con contraste: aspecto característico «sal y pimienta» en las imágenes ponderadas en T1.
TC con contraste: determinación del grado de invasión en los vasos, evaluación de la clasificación de Shamblin. Construcción de un modelo 3D para la planificación de la cirugía.
Angiografía: detección de la «señal de la lira» – divergencia de las arterias carótidas interna y externa. Permite evaluar la necesidad/posibilidad de embolización de esta zona como un paso anterior a la intervención quirúrgica.
PET-TC con 68Ga-DOTATATE: se utiliza si se sospechan focos múltiples o metastásicos.
Pruebas de laboratorio: determinación de niveles plasmáticos y urinarios de metanefrinas y normetanefrinas en caso de sospecha de tumor secretor.
Pruebas genéticas para detectar mutaciones en los genes SDH en caso de antecedentes familiares o tumores múltiples.
Encuentra más contenido científicamente preciso en nuestras redes sociales
Suscríbete y no te pierdas los últimos recursos
Tratamiento
El tratamiento del quimiodectoma carotídeo depende del tamaño del tumor, los síntomas clínicos, el riesgo de complicaciones, la actividad funcional, el perfil genético y el grado de Shamblin. Las principales modalidades de tratamiento son la escisión quirúrgica, la embolización preoperatoria y, en casos seleccionados, la radioterapia o la quimioterapia.
Embolización preoperatoria
La embolización preoperatoria se realiza para reducir la vascularización del tumor, disminuir el volumen de hemorragia intraoperatoria y facilitar la resección del tumor durante la cirugía.
Indicaciones:
tumores de grado II-III de Shamblin;
diámetro del tumor > 3 cm;
presencia de flujo sanguíneo arterial significativo (según TC/angiografía);
resección planificada con reconstrucción vascular.
Contraindicaciones:
tumores Shamblin I;
sin componente arterial (escasa vascularización);
anastomosis con circulación cerebral – riesgo de embolización cerebral.
Se realiza una angiografía selectiva de la arteria carótida externa. A través de un microcatéter se introducen agentes embolizantes: partículas de alcohol polivinílico (PVA), microesferas, menos comúnmente, pegamento o espirales. La angiografía de control confirma la reducción del flujo sanguíneo. La cirugía se realiza en las 24-48 horas siguientes a la embolización, mientras persista el efecto.
Tratamiento quirúrgico
Indicaciones:
tumores sintomáticos (dolor, disfagia, trastornos del habla);
crecimiento tumoral rápido;
tumores > 2,5–3 cm (incluso asintomáticos);
actividad confirmada por PET o bioquímica;
mutación SDHB confirmada (riesgo de malignidad);
edad < 60 años.
Tipos de tratamiento quirúrgico:
Resección extracapsular del tumor. Norma para Shamblin I-II, preservación vascular, riesgo mínimo.
Resección de la arteria carótida con reconstrucción. Más a menudo con Shamblin III. Tras la resección de la arteria carótida, se realiza una prótesis (Dacron, PTFE).
Neuromonitorización y microcirugía. Se utiliza cuando está cerca de los nervios craneales (IX-XII).
Posibles complicaciones:
hemorragia (especialmente con una embolización inadecuada);
daños en los nervios craneales (IX–XII): hasta un 30 % en los tumores grandes;
accidente cerebrovascular (cuando se deteriora el flujo sanguíneo colateral);
recurrencia (rara, con resección incompleta).
Quimioterapia
No es un tratamiento estándar para el quimiodectoma carotídeo, ya que la mayoría de los tumores son de crecimiento lento y naturaleza benigna. Sin embargo, en casos raros de curso maligno (progresión rápida, lesiones metastásicas) o en formas metastásicas inoperables, puede utilizarse terapia sistémica.
Terapia con radionúclidos ¹⁷⁷Lu-DOTATATO (PRRT)
Se prescribe en presencia de expresión de receptores de somatostatina mediante PET con ⁶⁸⁸Ga-DOTATATO.
FAQ
1. ¿La quimiodectoma carotidea es un tumor maligno?
En la mayoría de los casos — no. La quimiodectoma carotidea se considera un tumor benigno y de crecimiento lento. Sin embargo, con mutaciones en el gen SDHB, puede tener potencial maligno con metástasis.
2. ¿Se puede simplemente observar la quimiodectoma carotidea sin removerla?
Sí, en algunos casos es posible la vigilancia activa, especialmente si el tumor es pequeño ( 2,5 cm), asintomático, el paciente es anciano o tiene contraindicaciones para la cirugía. Sin embargo, con signos de crecimiento o compresión de las estructuras del cuello se requiere tratamiento.
3. ¿La operación para remover la quimiodectoma carotidea es peligrosa?
Los riesgos dependen del tamaño y ubicación del tumor. En formaciones grandes pueden presentarse complicaciones, incluyendo daño a los nervios craneales, hemorragia y eventos isquémicos. Para reducir los riesgos, frecuentemente se realiza una embolización preoperatoria.
4. ¿Se debe examinar a otros miembros de la familia si me diagnosticaron una quimiodectoma?
Si se encuentra una mutación hereditaria (por ejemplo, SDHD, SDHB), se recomienda asesoramiento genético y evaluación de los familiares cercanos, ya que la enfermedad puede ser familiar.
5. ¿Cómo distinguir la quimiodectoma carotidea de otros tumores del cuello?
La quimiodectoma carotidea generalmente se ubica en la bifurcación de la arteria carótida común, pulsa, se mueve horizontalmente, pero no verticalmente, y muestra el característico «signo de lira» en TC/RM. El diagnóstico se confirma mediante visualización y angiografía.
6. ¿Puede la quimiodectoma causar dolor o incomodidad?
En la mayoría de los casos — no, especialmente en las etapas iniciales. Sin embargo, a medida que crece el tumor, puede causar presión sobre los nervios y vasos, lo que lleva a dolor, ronquera, dificultad para tragar o mareos.
7. ¿Hay riesgo de recurrencia después de la extracción del tumor?
Con la extracción completa del tumor, el riesgo de recurrencia es bajo (menos del 5 %). Sin embargo, con una resección incompleta, forma hereditaria o presencia de mutación SDHB, puede haber recurrencia o desarrollo de nuevos focos. En tales casos, es importante la vigilancia a largo plazo.
Bibliografía
1.
Anatomía y Patología VOKA 3D: Atlas 3D completo de Anatomía y Patología [Internet]. Anatomía y Patología VOKA 3D
Disponible en: https://catalog.voka.io/
2.
Luna-Ortiz K, Reynoso-Noverón N, Herrera-Ponzanelli C, Favila-Lira S, Luna-Peteuil Z, Herrera-Gomez A, et al. Diferencias de sexo según la presentación étnica en tumores del cuerpo carotídeo: una revisión sistemática de la literatura. Int J Otorhinolaryngol Head Neck Surg. Junio de 2022; 8(6):527‑531. doi: 10.18203/issn.2454-5929.ijohns20221393.
3.
Butt N, Baek WK, Lachkar S, Iwanaga J, Mian A, Blaak C, et al. El cuerpo carotídeo y los tumores asociados: revisión actualizada con importancia clínica/quirúrgica. Br J Neurosurg. Octubre de 2019;33(5):500-503. doi: 10.1080/02688697.2019.1617404.
4.
Darouassi Y, Alaoui M, Mliha Touati M, Al Maghraoui O, En-Nouali A, Bouaity B, Ammar H. Tumores del cuerpo carotídeo: serie de casos y revisión de la literatura. Ann Vasc Surg. 2017 Ago;43:265-271. doi: 10.1016/j.avsg.2017.03.167.
5.
Gonzalez-Urquijo M, Castro-Varela A, Barrios-Ruiz A, Hinojosa-Gonzalez DE, Salas AKG, Morales EA, et al. Tendencias actuales en tumores del cuerpo carotídeo: revisión integral. Head Neck. Octubre de 2022;44(10):2316-2332. doi: 10.1002/hed.27147.
6.
Ozawa H. Manejo actual de los tumores del cuerpo carotídeo. Auris Nasus Larynx. Junio de 2024;51(3):501-506. doi: 10.1016/j.anl.2024.01.007.
7.
Piazza C, Lancini D, Tomasoni M, Zafereo M, Poorten VV, Hanna E, et al. Tumores malignos del cuerpo carotídeo: lo que sabemos, lo que hacemos y lo que necesitamos lograr. Una revisión sistemática de la literatura. Head Neck. 2024 Mar;46(3):672-687. doi: 10.1002/hed.27624.
Petersburg FL 33702, 7901 4th St N STE 300, ESTADOS UNIDOS
¡Gracias!
¡Tu mensaje ha sido enviado! Nuestros expertos se pondrán en contacto contigo en breve. Si tienes más preguntas, ponte en contacto con nosotros en info@voka.io.
Cookie Consent
Utilizamos cookies para mejorar su experiencia de navegación, analizar el tráfico del sitio y entregar contenido. Por favor, elija si acepta todas las cookies o desea rechazar el seguimiento no esencial.
Preferencias de Cookies
Gestione sus preferencias de cookies a continuación:
Las cookies esenciales permiten funciones básicas y son necesarias para el correcto funcionamiento del sitio web.
Nombre
Descripción
Duración
Configuración de Geolocalización
Esta cookie se utiliza para almacenar la configuración de consentimiento según la ubicación del visitante.
30 días
Preferencias de Cookies
Esta cookie se utiliza para almacenar las preferencias de consentimiento de cookies del usuario.
30 días
Google reCAPTCHA ayuda a proteger los sitios web del spam y el abuso verificando las interacciones de los usuarios mediante pruebas de seguridad.
Nombre
Descripción
Duración
_GRECAPTCHA
Google reCAPTCHA establece una cookie necesaria (_GRECAPTCHA) cuando se ejecuta con el propósito de proporcionar un análisis de riesgo.
179 días
Las cookies de estadísticas recopilan información de forma anónima. Esta información nos ayuda a entender cómo los visitantes utilizan nuestro sitio web.
Google Analytics es una herramienta poderosa que rastrea y analiza el tráfico del sitio web para ayudar a tomar decisiones de marketing fundamentadas.
Contiene información sobre la fuente de tráfico o la campaña que dirigió al usuario al sitio web. La cookie se establece cuando se carga el javascript GA.js y se actualiza cuando se envían datos al servidor de Google Analytics.
6 meses después de la última actividad
__utmv
Contiene información personalizada establecida por el desarrollador web a través del método _setCustomVar en Google Analytics. Esta cookie se actualiza cada vez que se envían nuevos datos al servidor de Google Analytics.
2 años después de la última actividad
__utmx
Utilizado para determinar si un usuario está incluido en una prueba A / B o multivariante.
18 meses
_ga
ID utilizado para identificar a los usuarios
2 años
_gali
Utilizado por Google Analytics para determinar qué enlaces en una página están siendo clicados
30 segundos
_ga_
ID utilizado para identificar a los usuarios
2 años
_gid
ID utilizado para identificar a los usuarios durante 24 horas después de la última actividad
24 horas
_gat
Utilizado para supervisar el número de solicitudes al servidor de Google Analytics al utilizar Google Tag Manager
1 minuto
_gac_
Contiene información relacionada con las campañas de marketing del usuario. Esta información se comparte con Google AdWords / Google Ads cuando las cuentas de Google Ads y Google Analytics están vinculadas.
90 días
__utma
ID utilizado para identificar usuarios y sesiones
2 años después de la última actividad
__utmt
Se utiliza para monitorear el número de solicitudes al servidor de Google Analytics
10 minutos
__utmb
Se utiliza para distinguir nuevas sesiones y visitas. Esta cookie se establece cuando se carga la biblioteca javascript GA.js y no existe una cookie __utmb. La cookie se actualiza cada vez que se envían datos al servidor de Google Analytics.
30 minutos después de la última actividad
__utmc
Se utiliza únicamente con versiones antiguas de Urchin de Google Analytics y no con GA.js. Se utilizaba para distinguir entre nuevas sesiones y visitas al final de una sesión.
Fin de sesión (navegador)
Clarity es un servicio de análisis web que realiza el seguimiento del tráfico del sitio web y genera informes sobre él del sitio web.