Chemodectoma (paraganglioma): etiologia, patogénese, classificação, diagnóstico, métodos de tratamento
Kizyukevich O.Cirurgião cardiovascular, MD
10 min ler·Maio 07, 2025
Este artigo é apenas para fins informativos
O conteúdo deste sítio Web, incluindo texto, gráficos e outros materiais, é fornecido apenas para fins informativos. Não se destina a servir de conselho ou orientação. Relativamente ao teu estado de saúde ou tratamento específico, consulta o teu profissional de saúde.
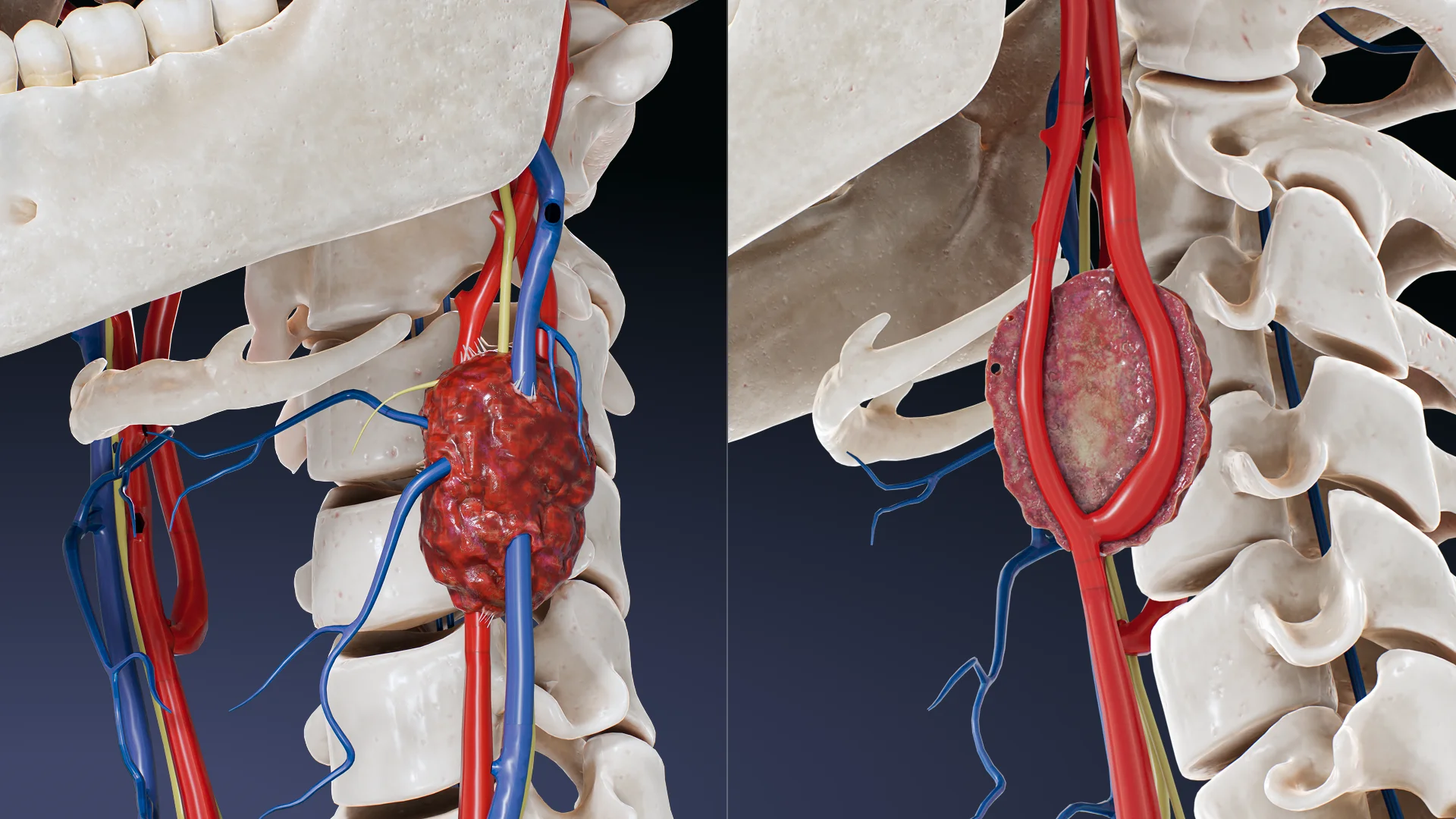
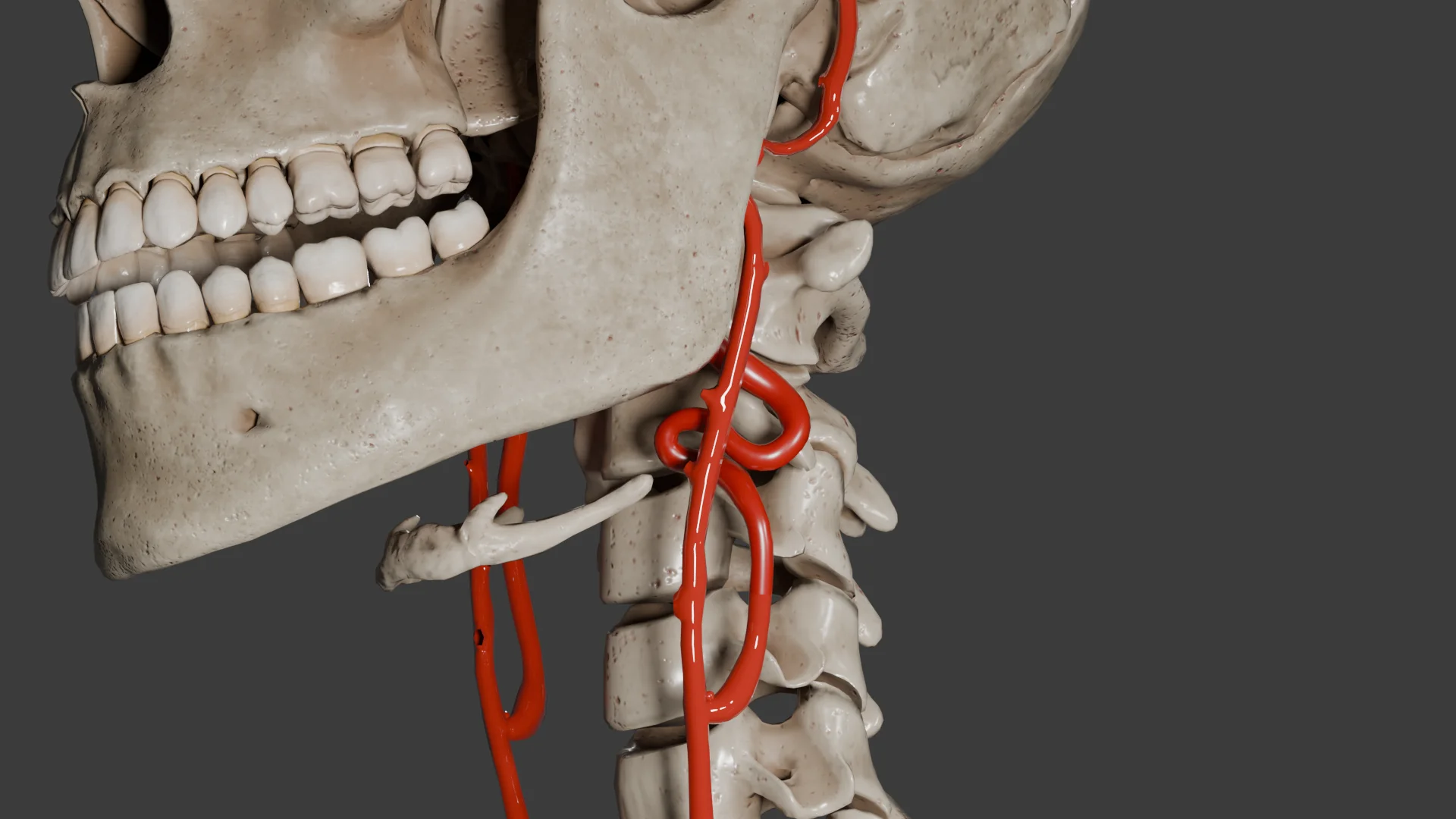
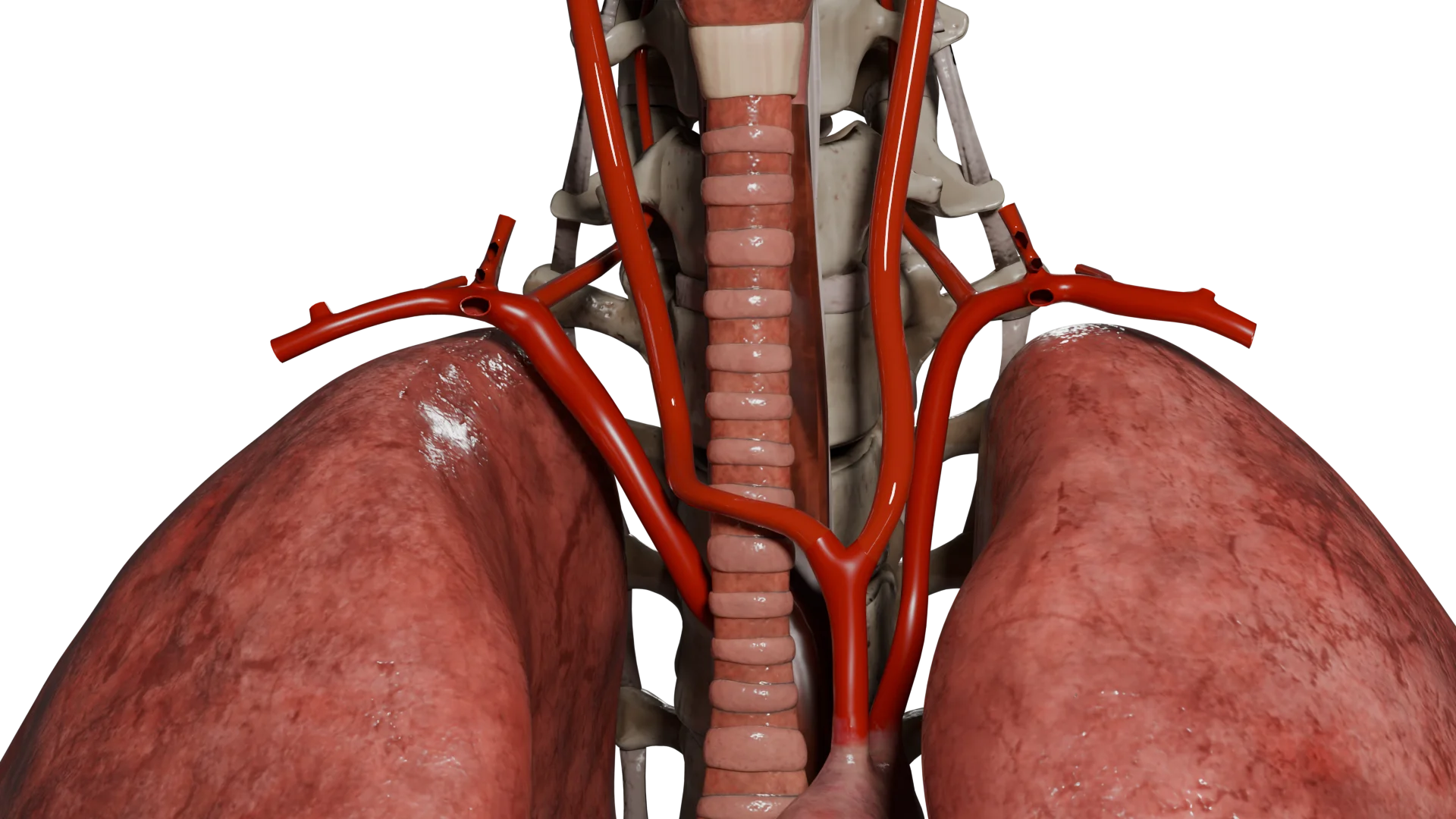
O paraganglioma da carótida (quimiodectoma) é um tumor raro, maioritariamente benigno, que surge das células quimiorreceptoras do corpo carotídeo localizado na bifurcação da artéria carótida comum.
Chemodectoma (paraganglioma)
Epidemiologia
Aproximadamente 1-2 casos por 100.000 habitantes.
As mulheres são mais frequentemente afectadas do que os homens.
A idade média do diagnóstico é de cerca de 45 anos.
A prevalência é elevada nos países da América Latina, especialmente no México, onde as mulheres representam até 90% dos casos.
Os habitantes de zonas de altitude elevada têm um risco acrescido de desenvolver quimiodectomas devido à hipóxia crónica.
A etiologia
Mutações genéticas: As mutações nos genes que codificam as subunidades do complexo succinato desidrogenase (SDH), que está envolvido na cadeia respiratória mitocondrial e no metabolismo celular, desempenham um papel importante. Os principais genes são:
SDHD – mais frequentemente associada a paragangliomas múltiplos da cabeça e do pescoço; transmitida por via paterna;
SDHB – associado a formas mais agressivas e a um maior risco de evolução maligna;
SDHC, SDHA e SDHAF2 estão menos frequentemente envolvidos, mas podem também contribuir para o desenvolvimento de tumores.
Síndromes associadas a quimiodectomas:
Síndrome de Von Hippel-Lindau (VHL);
Neoplasia endócrina múltipla tipo 2 (MEN2);
Neurofibromatose tipo 1 (NF1).
Hipóxia crónica: viver a > 2000 m acima do nível do mar, bem como condições como a doença pulmonar obstrutiva crónica e a doença cardíaca congénita, podem estimular a hiperplasia do corpo carotídeo.
História familiar: cerca de 10% dos casos são familiares.
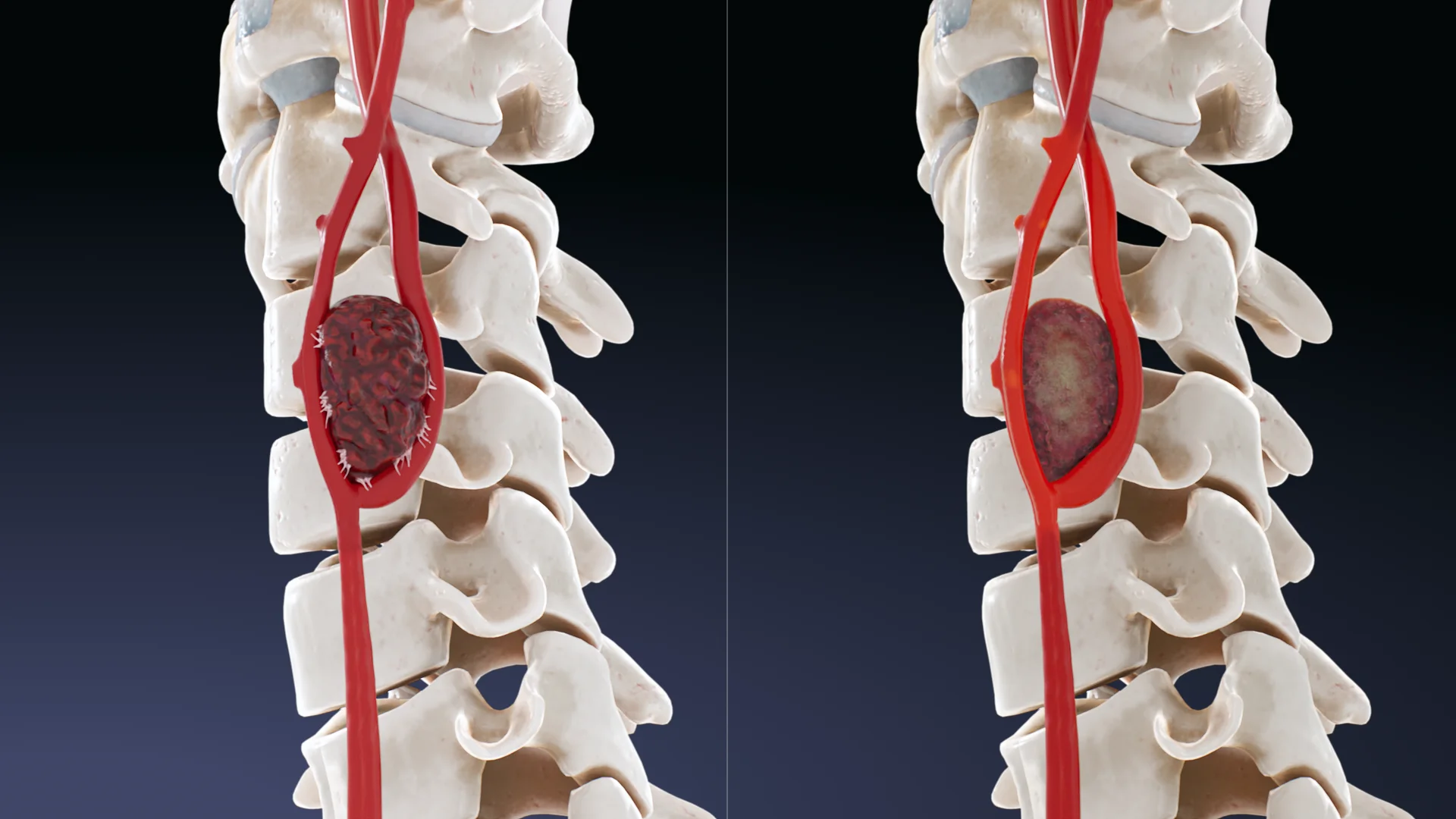
Animação 3D – Quimiodectoma tipo IAnimação 3D – Quimiodectoma tipo III
Patogénese
Mutações hereditárias (SDHx)
Mutações da SDH (enzima-chave do complexo II da cadeia respiratória mitocondrial e do ciclo de Krebs) → Acumulação de succinato (actua como “oncometabolito”) → Inativação das enzimas prolil hidroxilases (PHDs), controlando a degradação do HIF-1α (fator induzido pela hipóxia) → Acumulação de HIF-1α → Pseudo-hipóxia → Ativação de VEGF (angiogénese)/ GLUT1 (glicólise)/ PDGF (proliferação celular) → Transformação neoplásica das células quimiorreceptoras e formação de tumores.
Hipóxia crónica
Hipóxia (altitude, DPOC) → Diminuição da pO₂ → Ativação do recetor carotídeo → Hiperplasia celular → Acumulação de HIF-1α → Angiogénese e neoplasia.
Como resultado, o tumor aumenta gradualmente de tamanho. À medida que cresce, pode demonstrar um carácter localmente invasivo, a ponto de circundar ou comprimir estruturas anatómicas próximas, como as artérias carótidas interna e externa, os nervos vago, hioide e lingual, o que pode causar sintomas neurológicos e vasculares associados.
Classificação do paraganglioma
W.R. Shamblin propôs dividir os paragangliomas da carótida da panturrilha em três tipos:
Tipo I: tumores limitados até 3,5 cm, vagamente associados às paredes arteriais;
Tipo II: 3,5-5 cm, cobrem parcialmente as artérias carótidas, têm uma fusão mais densa com as paredes arteriais;
Tipo III: mais de 5 cm, cobrindo as artérias carótidas e/ou vasos próximos, os nervos têm uma fusão acentuadamente densa com estas estruturas.
Além disso, o quimiodectoma pode ser classificado de acordo com a etiologia: esporádico (até 85%), familiar (10-15%) e hiperplásico (1-5%).
Durante muito tempo, pode não haver sintomas. À medida que o tumor cresce, podem surgir queixas:
Tumor palpável: massa de crescimento lento, indolor e pulsátil na superfície lateral do pescoço (na zona do músculo esternoclavicular-papilar);
Sintomas neurológicos: rouquidão da voz, disfagia, dormência da língua causada pela compressão dos nervos cranianos (IX-XII);
Queixas relacionadas com a produção de catecolaminas (palpitações, ataques de hipertensão arterial, suores, dores de cabeça, tremores);
Raramente: tonturas, síncope com compressão do seio carotídeo.
Diagnóstico do paraganglioma
Exame físico: tumor denso e palpitante no pescoço, que se desloca horizontalmente, mas não verticalmente;
Ecografia com Doppler: tumor hipervascular na zona da bifurcação carotídea;
RM com contraste: aspeto caraterístico de “sal e pimenta” nas imagens ponderadas em T1;
TC com contraste: determinação do grau de invasão vascular, avaliação da classificação de Shamblin. Construção de um modelo 3D para o planeamento da cirurgia;
Angiografia: deteção do “sinal da lira” – divergência das artérias carótidas interna e externa. Permite avaliar a necessidade/possibilidade de embolização desta zona, como etapa prévia à intervenção cirúrgica;
PET-CT com 68Ga-DOTATATE: se houver suspeita de focos múltiplos ou metastáticos;
Exames laboratoriais: níveis plasmáticos e urinários de metanefrinas e normetanefrinas em caso de suspeita de tumor secretado;
Teste genético para detetar mutações nos genes SDH na presença de história familiar ou de tumores múltiplos.
Tratamento
O tratamento do quimiodectoma carotídeo depende do tamanho do tumor, dos sintomas clínicos, do risco de complicações, da atividade funcional, do perfil genético e do grau de Shamblin. As principais modalidades de tratamento são a excisão cirúrgica, a embolização pré-operatória e, em casos selecionados, a radioterapia ou a quimioterapia.
Embolização pré-operatória
Embolização pré-operatória para reduzir a vascularização do tumor e a perda de sangue intra-operatória, e para facilitar o isolamento do tumor durante a cirurgia.
Indicações:
Tumores de Shamblin de grau II-III;
Diâmetro do tumor >3 cm;
Presença de fluxo sanguíneo arterial significativo (de acordo com a TC/angiografia);
Ressecção planeada com reconstrução vascular.
Contra-indicações:
Tumores de Shamblin I;
Não tem componente arterial (pouca vascularização);
Anastomoses com a circulação cerebral – risco de embolização cerebral.
Procedimento: Realiza-se uma angiografia selectiva da artéria carótida externa. Os agentes embolizantes são injectados através de um microcateter: partículas de polivinilálcool (PVA), microesferas, menos frequentemente cola ou espirais. A angiografia de controlo confirma a redução do fluxo sanguíneo. A cirurgia é efectuada no prazo de 24-48 horas após a embolização, enquanto o efeito persistir.
Encontra mais conteúdos cientificamente exactos nas nossas redes sociais
Subscreve e não percas os recursos mais recentes
Tratamento cirúrgico
Indicações:
Tumores sintomáticos (dor, disfagia, perturbação da fala);
Crescimento rápido do tumor;
Tumores >2,5-3 cm (mesmo assintomáticos);
Atividade confirmada por PET ou bioquímica;
Mutação SDHB confirmada (risco de malignidade);
Idade <60 anos.
Tipos de tratamento cirúrgico:
Ressecção extracapsular do tumor. Padrão para Shamblin I-II, preservação vascular, risco mínimo;
Ressecção da artéria carótida com reconstrução. Mais frequentemente com Shamblin III. Após a ressecção da artéria carótida, é realizada uma prótese (Dacron, PTFE);
Neuromonitorização e microcirurgia. Utiliza-se quando está próximo dos nervos cranianos (IX-XII).
Possíveis complicações:
Hemorragia (especialmente em caso de embolização inadequada);
Danos nos nervos cranianos (IX-XII) – até 30% em tumores grandes;
Acidente vascular cerebral (quando o fluxo sanguíneo colateral é prejudicado);
Recidiva (rara, com ressecção incompleta).
Quimioterapia
Não se trata de um tratamento padrão para o quimiodectoma carotídeo, uma vez que a maioria dos tumores é de crescimento lento e de natureza benigna. No entanto, em casos raros de evolução maligna (progressão rápida, lesões metastáticas) ou em formas metastáticas inoperáveis, pode ser utilizada terapia sistémica.
Terapia com radionuclídeos ¹⁷⁷Lu-DOTATATE (PRRT)
Prescrito na presença de expressão de receptores de somatostatina por PET com ⁶⁸⁸Ga-DOTATATE.
FAQ
1. O quimiodectoma da carótida é um tumor maligno?
Na maioria dos casos, não o é. Os quimiodectomas da carótida são tumores benignos de crescimento lento. No entanto, na presença de mutações no gene SDHB, é possível um potencial maligno com metástases.
2. É possível observar apenas um quimiodectoma da carótida sem o remover?
Sim, a vigilância ativa é possível em alguns casos, especialmente se o tumor for pequeno (<2,5 cm), assintomático, idoso ou se houver contra-indicações para a cirurgia. No entanto, se houver sinais de crescimento ou compressão das estruturas do pescoço, é necessário tratamento.
3. A cirurgia para remover um quimiodectoma da carótida é perigosa?
Os riscos dependem do tamanho e da localização do tumor. No caso de grandes massas, são possíveis complicações, incluindo lesões dos nervos cranianos, hemorragias e eventos isquémicos. A embolização pré-operatória é frequentemente efectuada para reduzir os riscos.
4. Os outros membros da família têm de ser examinados se me for diagnosticado um quimiodectoma?
Se te for diagnosticada uma mutação hereditária (por exemplo, SDHD, SDHB), recomenda-se o aconselhamento genético e o rastreio dos teus familiares mais próximos, uma vez que a doença pode ser familiar.
5. Como se pode distinguir o quimiodectoma da carótida de outros tumores do pescoço?
O quimiodectoma carotídeo localiza-se geralmente na bifurcação da artéria carótida comum, é pulsátil, desloca-se horizontalmente, mas não verticalmente, e apresenta o caraterístico “sinal da lira” na TC/RM. O diagnóstico é esclarecido por imagiologia e angiografia.
6. Um quimiodectoma pode causar dor ou desconforto?
Na maioria dos casos, não, especialmente nas fases iniciais. No entanto, à medida que o tumor cresce, pode causar pressão nos nervos e vasos sanguíneos, resultando em dor, rouquidão, dificuldade em engolir ou tonturas.
7. Existe o risco de recidiva após a remoção do tumor?
Se o tumor for completamente removido, o risco de recorrência é baixo (menos de 5%). Contudo, em caso de ressecção incompleta, forma hereditária ou presença de mutação SDHB, é possível a recorrência ou o desenvolvimento de novos focos. Nestes casos, é importante um acompanhamento a longo prazo.
Lista de fontes
1.
Catálogo VOKA.
https://catalog.voka.io/
2.
Luna-Ortiz, K., Reynoso-Noverón, N., Herrera-Ponzanelli, C., Favila-Lira, S., Luna-Peteuil, Z., Herrera-Gomez, A., & Gacia-Ortega, D. Y. Sex differences according to ethnic presentation in carotid body tumors: a systematic literature review. International Journal of Otorhinolaryngology and Head and Neck Surgery. 2022 Jun;8(6):527–531. DOI:10.18203/issn.2454-5929.ijohns20221393.
3.
Butt N, Baek WK, Lachkar S, Iwanaga J, Mian A, Blaak C, Shah S, Griessenauer C, Tubbs RS, Loukas M. The carotid body and associated tumors: updated review with clinical/surgical significance. Br J Neurosurg. 2019 Oct;33(5):500-503. doi: 10.1080/02688697.2019.1617404.
4.
Darouassi Y, Alaoui M, Mliha Touati M, Al Maghraoui O, En-Nouali A, Bouaity B, Ammar H. Carotid Body Tumors: A Case Series and Review of the Literature. Ann Vasc Surg. 2017 Aug;43:265-271. doi: 10.1016/j.avsg.2017.03.167.
5.
Gonzalez-Urquijo M, Castro-Varela A, Barrios-Ruiz A, Hinojosa-Gonzalez DE, Salas AKG, Morales EA, González-González M, Fabiani MA. Current trends in carotid body tumors: Comprehensive review. Head Neck. 2022 Oct;44(10):2316-2332. doi: 10.1002/hed.27147.
6.
Ozawa H. Current management of carotid body tumors. Auris Nasus Larynx. 2024 Jun;51(3):501-506. doi: 10.1016/j.anl.2024.01.007.
7.
Piazza C, Lancini D, Tomasoni M, Zafereo M, Poorten VV, Hanna E, Mäkitie AA, Fernandez-Alvarez V, Kowalski LP, Chiesa-Estomba C, Ferlito A. Malignant carotid body tumors: What we know, what we do, and what we need to achieve. A systematic review of the literature. Head Neck. 2024 Mar;46(3):672-687. doi: 10.1002/hed.27624.
São Petersburgo FL 33702, 7901 4th St N STE 300, EUA
Obrigado!
A tua mensagem foi enviada! Os nossos especialistas entrarão em contacto contigo em breve. Se tiveres mais perguntas, contacta-nos através de info@voka.io

.webp)


/aortic%20dissection_main.webp)
