Restrictive Cardiomyopathy: Etiology, Pathogenesis, Clinical Manifestations, Diagnosis, and Treatment
Oleg K.Cardiovascular surgeon, MD
12 min read·December 23, 2025
This article is for informational purposes only
The content on this website, including text, graphics, and other materials, is provided for informational purposes only. It is not intended as advice or guidance. Regarding your specific medical condition or treatment, please consult your healthcare provider.
Restrictive cardiomyopathy (RCM) is a rare type of primary or secondary cardiomyopathy. It is defined by impaired diastolic filling of the left and/or right ventricle, while systolic function and wall thickness are normal or only mildly abnormal. The myocardium becomes stiff, which raises diastolic pressures and ultimately leads to congestive heart failure.
RCM requires careful differential diagnosis because it can be caused by many processes, ranging from genetic disorders to systemic diseases.
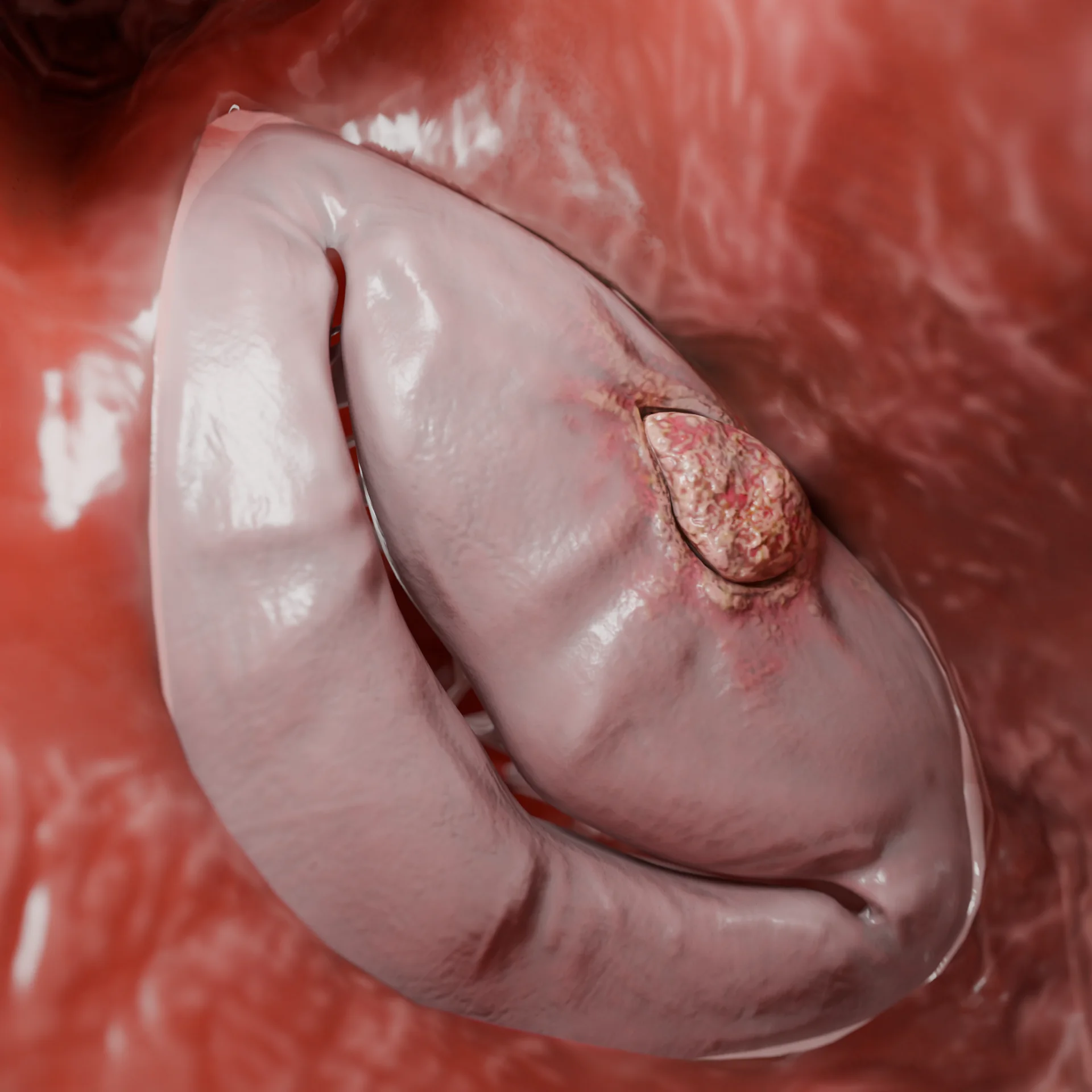
Left Ventricular Cavity in Restrictive Cardiomyopathy Secondary to Endomyocardial Fibrosis — 3D Model
Etiology of Restrictive Cardiomyopathy
RCM may be idiopathic (primary) or secondary to systemic or infiltrative diseases. The main etiologic groups include:
Infiltrative diseases:
Amyloidosis (AL and ATTR forms): This is the most common cause of RCM in adults.
Sarcoidosis: This involves granulomatous infiltration of the myocardium, which leads to increased stiffness and conduction abnormalities.
Storage diseases:
Fabry disease: This results from deficiency of the enzyme α-galactosidase A and leads to progressive myocardial involvement.
Hemochromatosis: In this condition, iron is deposited within the myocardium, which over time leads to fibrosis and a restrictive physiology.
Radiation- and chemotherapy-induced injury:
Anthracycline cardiotoxicity (e.g., doxorubicin).
Mediastinal radiotherapy.
Endomyocardial disease:
Endomyocardial fibrosis: This condition involves fibrotic thickening of the endocardium and can lead to a restrictive pattern of cardiomyopathy.
Loeffler endocarditis (hypereosinophilic endomyocardial disease): This condition is caused by eosinophil-mediated toxic injury to the endocardium and myocardium, followed by fibrosis.
In some patients, no clear cause can be identified. In such cases, the condition is classified as idiopathic restrictive cardiomyopathy.
Pathogenesis and Classification
The key pathophysiologic feature of RCM is reduced myocardial compliance (distensibility), with preserved or only mildly reduced systolic function. The ventricular walls become rigid and cannot relax properly during diastole.
This initiates a cascade of pathological changes:
Elevated filling pressures: Stiff ventricles require higher pressures to fill. As a result, venous congestion develops in both the pulmonary and systemic circulation.
Reduced cardiac output: Because diastolic filling is restricted, both stroke volume and cardiac output fall, even when the ejection fraction is still normal.
Atrial dysfunction: Persistently high filling pressures in the ventricles are transmitted back to the atria. Over time, this leads to marked atrial dilation, a reduced atrial “boost” to ventricular filling, and worsening symptoms of heart failure.
Fibrosis and structural remodeling: Chronic pressure overload and ongoing injury promote myocardial fibrosis and structural remodeling. This further reinforces the restrictive phenotype.
Arrhythmias: Atrial fibrillation, atrioventricular block, and bradyarrhythmias are common, especially in amyloidosis and sarcoidosis, where the conduction system is involved. In addition, the combination of atrial dilation and arrhythmias increases the risk of intracardiac thrombus and thromboembolism.
In advanced stages, these mechanisms culminate in refractory diastolic heart failure with low cardiac output, poor response to standard heart-failure therapy, and a high risk of thromboembolic complications.
Pathogenesis in Specific Forms of Restrictive Cardiomyopathy
The detailed mechanisms depend on the underlying disease:
Amyloidosis: Extracellular deposition of amyloid between cardiomyocytes causes wall thickening, stiffness, fibrosis, and involvement of the conduction system.
Fabry disease: Intracellular accumulation of glycosphingolipids within cardiomyocytes leads to progressive myocyte dysfunction.
Sarcoidosis: Granulomatous inflammation is gradually replaced by patchy fibrosis, which predisposes to conduction block and ventricular arrhythmias.
Classification of Restrictive Cardiomyopathy
Type of RCM
Description
Primary (idiopathic)
Disease confined to the myocardium; the underlying cause is unknown.
Infiltrative
Deposition of abnormal substances such as amyloid in the interstitial space.
Storage diseases
Intracellular accumulation of metabolites (e.g., iron, glycogen, lipids).
Endomyocardial
Fibrosis of the endocardium and inner myocardium (e.g., Loeffler endocarditis).
Radiation-induced and drug-induced
Myocardial damage due to ionizing radiation and/or cardiotoxic chemotherapy.
Clinical manifestations
In RCM, diastolic dysfunction and venous congestion are the predominant features. Typical symptoms and signs include:
Dyspnea: initially on exertion due to impaired left ventricular filling, later also at rest.
Orthopnea: breathlessness when lying flat and episodes of pulmonary edema related to pulmonary venous congestion.
Signs of systemic congestion: jugular venous distension, peripheral edema (especially of the legs), and ascites.
Symptoms of low cardiac output: marked fatigue, reduced exercise tolerance, weakness, orthostatic hypotension, and episodes of syncope.
Arrhythmias: atrial fibrillation is common and often worsens symptoms and prognosis. Other atrial or ventricular arrhythmias may also occur.
Thromboembolic events: the risk of stroke and other embolic complications is increased because of blood stasis in markedly enlarged atria and coexisting arrhythmias.
Diagnosis
The diagnosis of RCM requires a comprehensive approach. It is important both to exclude constrictive pericarditis and to identify the underlying cause.
Laboratory tests
NT-proBNP or BNP: These biomarkers are usually elevated and reflect the severity of heart failure, although they are not specific for RCM.
Cardiac troponins: These may be mildly elevated in infiltrative forms (such as amyloidosis), even in the absence of overt ischemia.
Screening tests: These may include serum amyloid A; serum protein electrophoresis and immunoglobulin studies to exclude AL amyloidosis and other paraproteinemias; iron and ferritin levels to assess for hemochromatosis; and measurement of α-galactosidase A (GLA) activity in suspected Fabry disease.
Instrumental methods
1. Echocardiography Echocardiography is central to the diagnosis of RCM and often shows a characteristic pattern:
Increased ventricular wall thickness with normal or reduced cavity size.
Marked enlargement of both atria, especially the left atrium.
Reduced ventricular compliance with a restrictive pattern of diastolic filling (high E/A ratio and shortened deceleration time).
Preserved ejection fraction, which usually falls only in late stages.
In cardiac amyloidosis: an “apical sparing” pattern on longitudinal strain imaging, with reduced strain in basal and mid segments and relative preservation of the apex.
2. Electrocardiography (ECG) RCM on ECG often shows low-voltage QRS complexes, atrial fibrillation, atrioventricular block, and ventricular arrhythmias.
3. Cardiac MRI with gadolinium (late gadolinium enhancement) Cardiac MRI provides detailed information on myocardial infiltration and fibrosis:
Subendocardial or diffuse late gadolinium enhancement is typical of amyloidosis.
Patchy areas of enhancement are suggestive of cardiac sarcoidosis.
In idiopathic RCM, a specific enhancement pattern may be absent.
Cardiac catheterization is used to measure pressures within the cardiac chambers. Analysis of ventricular pressure tracings and their respiratory variation helps distinguish RCM from constrictive pericarditis, which is crucial for choosing the appropriate treatment strategy.
5. Additional investigations
Endomyocardial biopsy: This invasive procedure carries risk but can provide a definitive diagnosis in infiltrative, inflammatory, or storage diseases.
99mTc-PYP (pyrophosphate) scintigraphy: This is a sensitive method for detecting transthyretin (ATTR) cardiac amyloidosis. It also helps distinguish ATTR from AL amyloidosis.
FDG-PET: This imaging modality helps identify active inflammatory lesions in cardiac sarcoidosis.
Cardiac CT: This is useful for assessing pericardial calcification and supports the diagnosis of constrictive pericarditis when it is suspected.
Endomyocardial Fibrosis Causing Restrictive Cardiomyopathy — 3D Model
Find more scientifically accurate content on our social media
Subscribe and don’t miss out the latest resources
Treatment of Restrictive Cardiomyopathy
Current recommendations distinguish between symptomatic therapy and disease-specific treatment directed at the underlying cause.
Medication therapy
Loop diuretics (e.g., furosemide, torsemide): used to relieve symptoms of congestion. They reduce pulmonary and systemic venous pressure but must be titrated carefully to avoid an excessive fall in preload and cardiac output.
Beta-blockers and non-dihydropyridine calcium channel blockers: used to control heart rate in tachycardia and atrial fibrillation. In cardiac amyloidosis they should be used with caution, as some patients tolerate them poorly.
Anticoagulants: indicated in atrial fibrillation to prevent thrombus formation and systemic embolism.
Disease-specific therapy
ATTR amyloidosis: treatment includes tafamidis, which stabilizes transthyretin and slows progression of cardiac involvement.
AL amyloidosis: therapy is based on chemotherapy regimens (for example, bortezomib-based combinations with cyclophosphamide) and, in selected patients, bone-marrow or stem-cell transplantation.
Fabry disease: pharmacologic chaperone therapy (migalastat) and enzyme replacement therapy (agalsidase alfa or agalsidase beta).
Sarcoidosis: glucocorticoids (such as prednisolone) and, when needed, additional immunosuppressive agents (e.g., methotrexate or azathioprine).
Hemochromatosis: iron chelation therapy (e.g., deferoxamine).
Surgical Therapy
Pacemaker implantation: indicated in significant bradycardia or high-grade atrioventricular block.
Implantable cardioverter-defibrillator (ICD): used for prevention of sudden cardiac death, particularly in patients with documented ventricular tachyarrhythmias or extensive myocardial scarring, such as in cardiac sarcoidosis.
Heart transplantation: considered in severe, refractory heart failure in suitable candidates, especially when disease-specific treatment is ineffective or when the underlying systemic disease is controlled.
FAQ
1. What is restrictive cardiomyopathy and how does it differ from other types of cardiomyopathy?
Restrictive cardiomyopathy (RCM) is a condition in which the myocardium becomes stiff and loses its ability to relax normally during diastole. As a result, the ventricles fill poorly, even though systolic function and chamber size are often near normal. By contrast, in dilated cardiomyopathy the chambers are enlarged and systolic function is typically reduced.
2. What symptoms are typical of restrictive cardiomyopathy?
The main symptoms are related to congestion and reduced filling of the heart. Patients often report breathlessness, rapid fatigue, peripheral edema, visible neck-vein distension, and ascites. These features reflect increased pressures in the pulmonary and systemic venous systems due to diastolic dysfunction.
3. Can restrictive cardiomyopathy be hereditary?
Yes. Yes. Some forms of RCM have a genetic basis. A typical example is variant transthyretin amyloidosis (ATTRv), in which a mutation in the transthyretin gene leads to progressive amyloid deposition in the heart and other organs.
4. Can restrictive cardiomyopathy be completely cured?
In most patients, RCM is a chronic, progressive disease. However, if a potentially reversible cause is found early (e.g., sarcoidosis or eosinophilic myocarditis), targeted treatment may stabilize the condition and, in some cases, lead to improvement.
5. How is restrictive cardiomyopathy diagnosed?
Diagnosis of RCM is based on a combination of imaging, ECG, biomarkers, and laboratory tests. Echocardiography is central, and is often supplemented by cardiac CT or MRI.
Additional tools include ECG, measurement of NT-proBNP and troponins, endomyocardial biopsy in selected cases, nuclear scintigraphy, and focused laboratory studies to detect amyloidosis and other underlying diseases.
6. Which diseases most commonly lead to restrictive cardiomyopathy?
The most frequent causes are amyloidosis (ATTR and AL types), sarcoidosis, and hemochromatosis.
RCM may also be associated with systemic connective-tissue diseases, eosinophilic endomyocarditis, and radiation-induced heart disease.
7. What treatments are used in restrictive cardiomyopathy?
Management has two main components. First, treatment should target the underlying cause whenever possible (e.g., chemotherapy in AL amyloidosis). Second, symptomatic therapy is used to control heart failure and arrhythmias. This may include diuretics, drugs for rhythm and rate control, anticoagulation, and, in advanced stages, consideration of heart transplantation.
8. What is the prognosis in restrictive cardiomyopathy?
Prognosis depends largely on the etiology and how early the disease is recognized. AL amyloidosis is generally associated with a poor prognosis. By contrast, some patients with ATTR amyloidosis may have a more stable course, especially when treated with targeted agents.
9. Is physical exercise allowed in patients with restrictive cardiomyopathy?
Physical activity is usually restricted. Most patients are advised to limit themselves to light, low-intensity exercise that does not provoke symptoms. Any training plan should be discussed with a cardiologist, and unsupervised intense exercise is generally discouraged.
10. When is heart transplantation indicated in restrictive cardiomyopathy?
Heart transplantation is considered in end-stage heart failure due to RCM when symptoms persist despite optimal medical and device therapy. Candidates should have no major extracardiac organ damage and no absolute contraindications to transplantation. Transplantation is particularly relevant when disease-specific treatment (e.g., therapy for AL amyloidosis) is ineffective or no longer feasible.
References
1.
VOKA Catalog. [Electronic resource].
https://catalog.voka.io/
2.
Rapezzi, C., Aimo, A., Barison, A., et al. (2022). Restrictive cardiomyopathy: Definition and diagnosis. European Heart Journal, 43(45), 4679–4693. https://doi.org/10.1093/eurheartj/ehac543
3.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi: 10.1093/eurheartj/ehad194.
4.
Muchtar, E., Blauwet, L. A., & Gertz, M. A. (2017). Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circulation Research, 121(7), 819–837. 2017 Sep 15;121(7):819-837. https://doi.org/10.1161/CIRCRESAHA.117.310982
5.
Vio, R., Angelini, A., Basso, C., et al. (2021). Hypertrophic cardiomyopathy and primary restrictive cardiomyopathy: Similarities, differences and phenocopies. Journal of Clinical Medicine, 10(9), 1954. J Clin Med. 2021 May 1;10(9):1954. https://doi.org/10.3390/jcm10091954
6.
Garcia, M. J. (2016). Constrictive pericarditis versus restrictive cardiomyopathy? J Am Coll Cardiol. Journal of the American College of Cardiology, 67(17), 2061–2076. https://doi.org/10.1016/j.jacc.2016.01.076 doi: 10.1016/j.jacc.2016.01.076.
7.
Arbeláez-Cortés, Á., Quintero-González, D. C., Cuesta-Astroz, Y., et al. (2020). Restrictive cardiomyopathy in a patient with systemic sclerosis and Fabry disease: A case-based review. Rheumatology International, 40(3), 489–497. 2020 Mar;40(3):489-497. https://doi.org/10.1007/s00296-019-04453-y