Ependymomas: Etiology, Classification, Diagnosis, Treatment, and Prognosis
Svetlana D.Surgical oncologist, MD
17 min read·November 13, 2025
This article is for informational purposes only
The content on this website, including text, graphics, and other materials, is provided for informational purposes only. It is not intended as advice or guidance. Regarding your specific medical condition or treatment, please consult your healthcare provider.
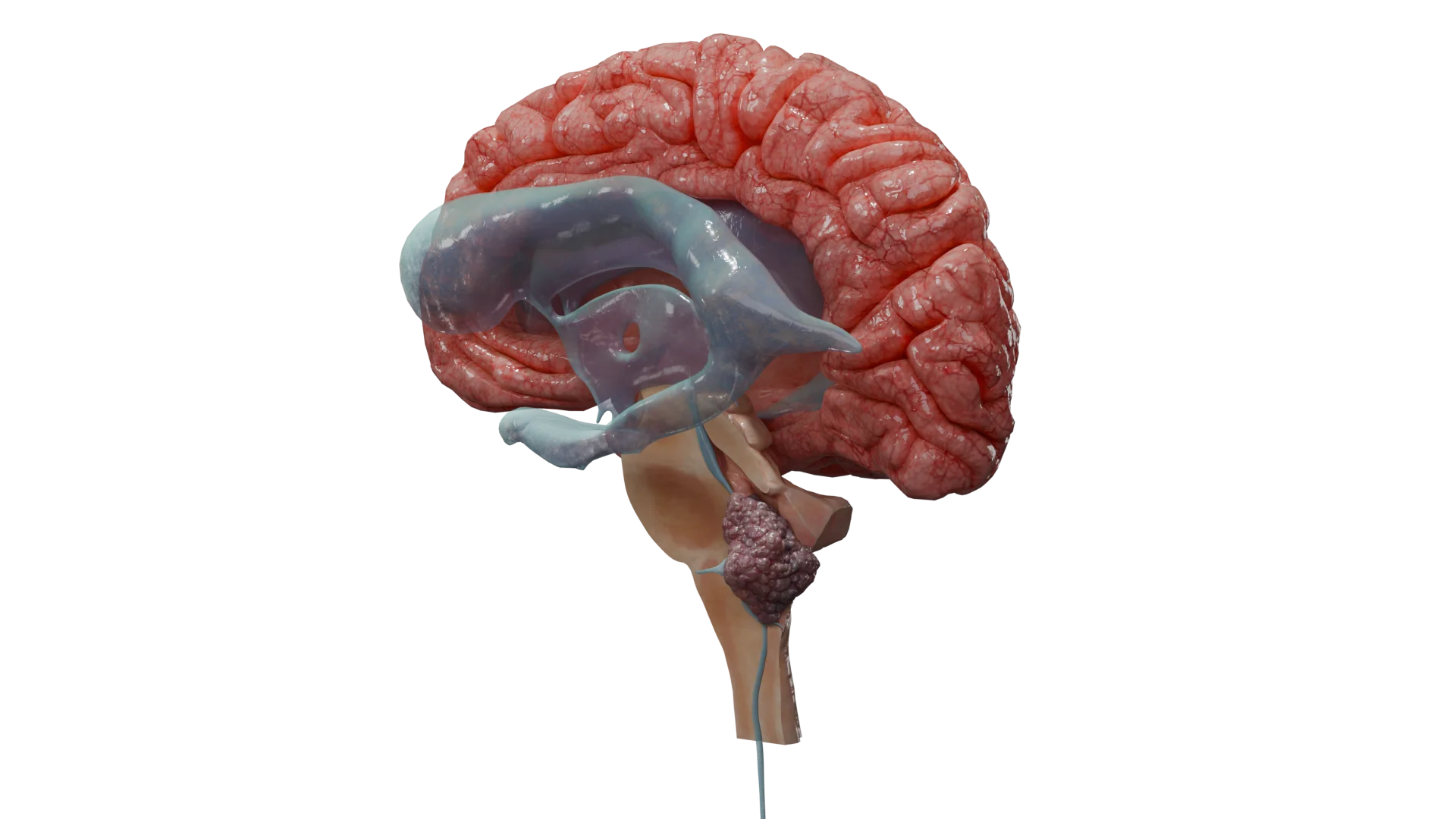
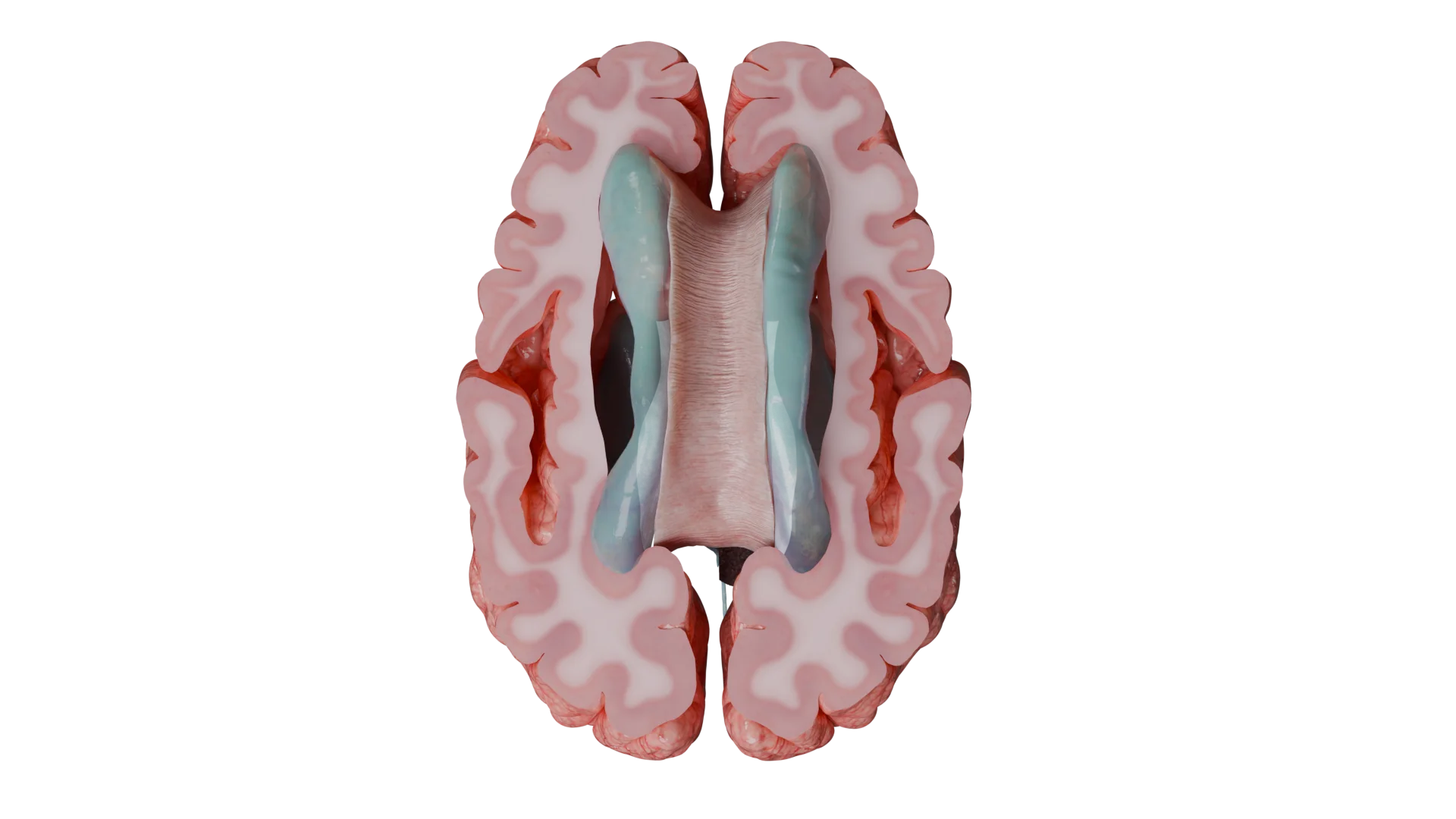
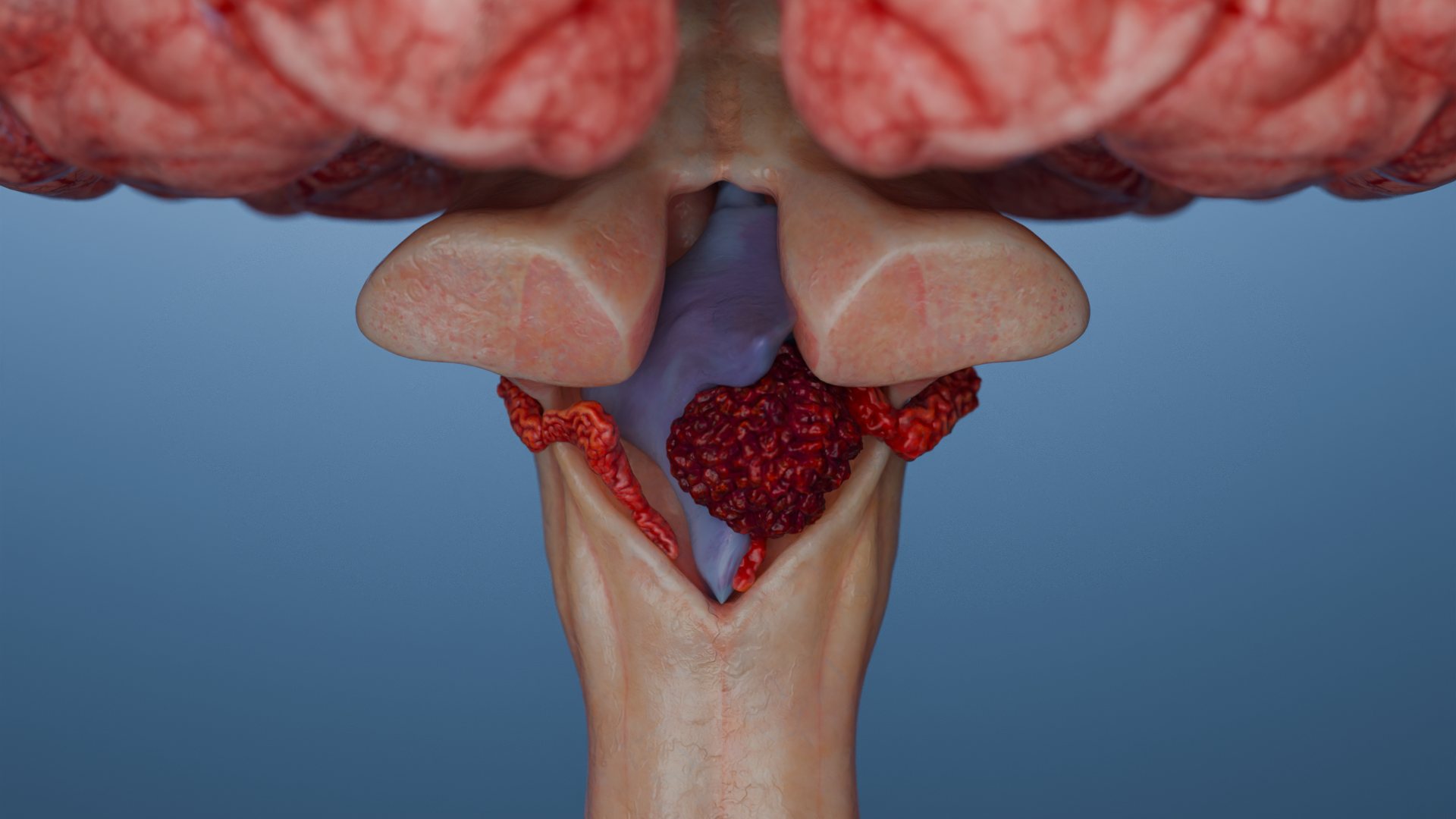
Ependymomas are a group of glial tumors arising from ependymal cells that line the ventricular system of the brain and the central canal of the spinal cord. These tumors can occur throughout the central nervous system (CNS), most commonly in the posterior fossa and spinal cord. The current standard of care for ependymomas emphasizes maximal surgical resection followed by adjuvant radiotherapy. Prognosis depends on the anatomical location, histological features, and molecular-genetic profile of the tumor.
The precise etiology of ependymomas remains unclear.
However, several mechanisms are implicated:
Genetic alterations: Common findings include structural DNA changes such as chromosomal imbalances (e.g., gain of chromosome 1q), gene fusions (e.g., ZFTA-RELA), deletions, and mutations that activate oncogenic pathways and disrupt cell growth regulation.
Epigenetic changes (altered gene activity): Disruption of DNA and histone methylation is particularly important. For example, loss of H3K27me3 trimethylation in PF-EPN-A subtypes suppresses gene expression involved in cellular differentiation.
Hereditary factors: Some spinal ependymomas are associated with NF2-related schwannomatosis.
Thus, ependymomas result from a complex interplay of genetic and epigenetic disruptions that lead to loss of cellular control and excessive proliferation.
Epidemiology
Ependymomas can be diagnosed at any age, with peak incidence in early childhood (median age: 5 years).
In pediatric cases, 90 % of tumors are intracranial, predominantly located in the posterior fossa.
In adults, the overall incidence is lower, with approximately 65 % of cases occurring in the spinal cord.
Clinical manifestations
Ependymoma symptoms vary by tumor location:
Supratentorial tumors (superior to the tentorium cerebelli): seizures, focal neurologic deficits (e.g., speech disturbances, personality changes, memory and attention impairment, hemiparesis), and signs of increased intracranial pressure (headache, nausea, vomiting, papilledema).
Supratentorial tumors typically develop within the lateral ventricles.
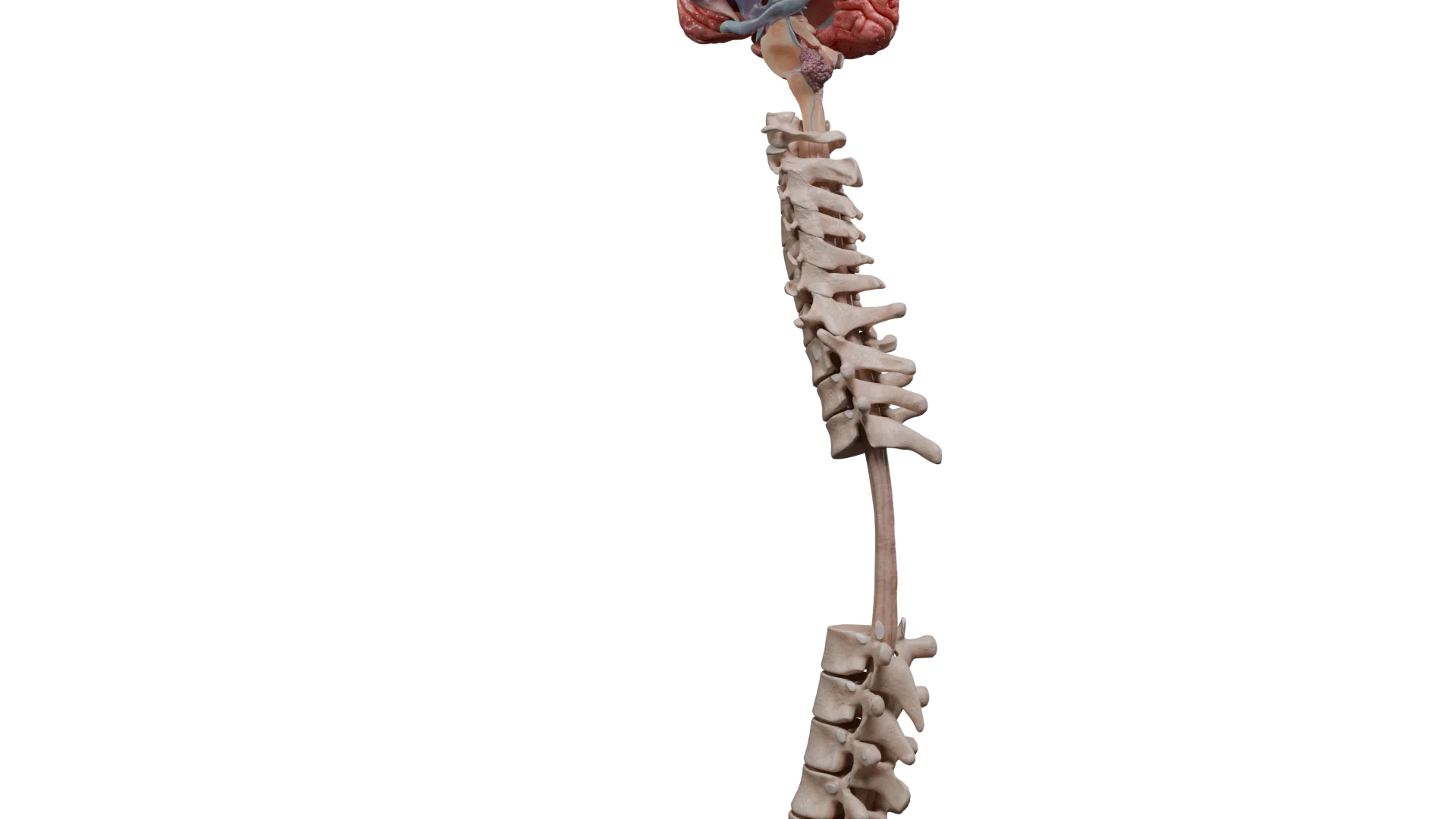
Spinal ependymomas: back pain, radicular pain, progressive muscle weakness, and pelvic organ dysfunction (bladder or bowel incontinence; occasionally, erectile dysfunction).
Location of Spinal Ependymomas
Classification of Ependymomas
Ependymomas are classified based on anatomical location, histological features, and molecular markers.
In 2021, the World Health Organization (WHO) introduced significant updates to CNS tumor classification, incorporating molecular markers that enhance prognostic accuracy and support personalized treatment strategies.
These updates also refine the conventional histological classification of ependymomas.
WHO 2021 Classification of Ependymomas Incorporating Molecular Features
The WHO 2021 classification of CNS tumors defines 10 types of ependymomas:
Supratentorial (ST-EPN):
Supratentorial ependymoma, not otherwise specified/not elsewhere classified (ST-EPN-NOS/NEC);
ZFTA fusion-positive supratentorial ependymoma;
YAP1 fusion-positive supratentorial ependymoma.
Posterior fossa ependymoma (PF-EPN):
Posterior fossa ependymoma, not otherwise specified/not elsewhere classified (PF-EPN-NOS/NEC);
Posterior fossa ependymoma, group А;
Posterior fossa ependymoma, group B.
Spinal ependymoma (SP-EPN):
Spinal ependymoma;
MYCN-amplified spinal ependymoma.
Myxopapillary ependymoma (MEPN). Most often found within the caudal portion of the spinal cord.
Subependymoma (SubEPN). Most often found within the fourth ventricle and lateral ventricles.
Rare; favorable prognosis; typically occurs in infants
Supratentorial ependymoma, not otherwise specified (ST-EPN-NOS/NEC)
2 or 3
Heterogeneous mutations, not elsewhere classified (NEC) or not otherwise specified (NOS) in molecular terms
Heterogeneous group, prognosis and therapy require further research
Posterior fossa ependymoma, group А (PF-EPN-A)
–
H3K27me3 loss; often 1q+
Aggressive; poor prognosis; most common in infants and young children
Posterior fossa ependymoma, group B (PF-EPN-B)
–
Chromosomal instability; H3K27me3 preserved
Favorable prognosis; older patients (adolescents and adults)
Posterior fossa ependymoma, not otherwise specified (PF-EPN-NOS/NEC)
2 or 3
Morphologically, posterior fossa ependymoma, not elsewhere classified (NEC) or not otherwise specified (NOS) in molecular terms
Diagnosed by location and typical histological signs; prognosis depends on extent of resection and clinical course
Spinal ependymoma
2 or 3
No consistent molecular markers; often chromosome 22q loss involving the NF2 gene
Intermediate prognosis; surgery + radiotherapy
MYCN-amplified spinal ependymoma (SP-EPN-MYCN)
–
MYCN amplification
Highly aggressive; poor prognosis
Myxopapillary ependymoma (MEPN)
2
Variable clinical presentation in adults and children
Most often found in the caudal spinal cord; local recurrence; recovery is possible following surgery and radiotherapy
Subependymoma (SubEPN)
1
Typically lacks aggressive mutations; benign; occasionally TERT mutation
Any portion of the ventricular system and spinal cord; slow progression; often incidental finding; favorable prognosis
WHO Malignancy Grade Definition (WHO Grade)
By malignancy, ependymomas are classified as WHO Grade II–III. The higher the WHO grade, the more aggressive the tumor and the poorer the patient’s prognosis.
According to the updated 2021 WHO classification of CNS tumors, no grade is assigned to newly defined molecular subtypes of ependymoma, such as YAP1-fusion positive (ST-EPN-YAP1), ZFTA-fusion positive (ST-EPN-ZFTA), PF-A and PF-B. Tumor grading (grade 1, 2, or 3, indicating degree of malignancy) applies only to classic ependymomas, including those defined morphologically but lacking molecular designation and referred to as “NEC/NOS”.
Definitions of NOS и NEC:
NOS (Not Otherwise Specified) — «Unspecified»: This term is used when a tumor has not undergone (or cannot undergo) the full panel of molecular/genetic or other ancillary tests, and the diagnosis is based solely on primary (morphological) criteria. For example, in the case of ependymoma: if a brain tumor exhibits classic histological features of ependymoma but molecular subgrouping (e.g., ZFTA-fusion, YAP1-fusion, PF-A, PF-B) has not been performed or the analysis is inconclusive, the diagnosis is recorded as “supratentorial ependymoma, NOS” or “posterior fossa ependymoma, NOS”.
NEC (Not Elsewhere Classified) — «Unclassifiable by current standards»: This term applies when a tumor has been fully characterized using contemporary diagnostic methods but does not fit into any of the recognized, standardized subtypes or groups. If the tumor meets the histological and molecular criteria for ependymoma and has undergone comprehensive evaluation, yet does not match any of the described molecular subtypes (e.g., ZFTA, YAP1, PF-A, PF-B), the diagnosis is rendered as “ependymoma, NEC”.
Ependymomas can metastasize within the CNS by disseminating neoplastic cells through cerebrospinal fluid circulation.
Diagnosis of Ependymomas
Magnetic Resonance Imaging (MRI)
The cornerstone of diagnosis is contrast-enhanced MRI of the brain and spinal cord.
Supratentorial and infratentorial ependymomas typically exhibit calcifications and cystic components; they are also characterized by hemorrhages and heterogeneous MR-signal enhancement.
On imaging, ependymomas are generally hypointense on T1-weighted images and hyperintense on T2-weighted images, often demonstrating marked contrast enhancement.
Spinal ependymomas are less frequently calcified and may show T2 hypointensity due to hemosiderin deposition — a feature known as the “cap sign”.
Compared to other spinal cord tumors, myxopapillary ependymomas usually present as T1-isointense lesions and T2-hyperintense lesions.
Cerebrospinal Fluid Cytology
Cerebrospinal fluid (CSF) cytology is important for staging and is most often performed postoperatively, including to help define the scope of adjuvant radiotherapy.
Histologic examination
Histological confirmation at the time of tumor resection is mandatory. A tumor specimen obtained during surgery undergoes microscopic examination to determine its type and grade of malignancy.
Surgical resection is the primary treatment modality for ependymomas.
Moreover, maximal tumor removal is critical for improving prognosis. Due to the proximity of vital structures, surgery requires a high level of expertise.
In cases of hydrocephalus, shunting procedures may be performed to prevent excessive CSF accumulation in the cerebral ventricles.
Due to the typical location of ependymomas, complete surgical resection is often challenging and associated with a high risk of complications. In such cases, postoperative radiotherapy becomes increasingly necessary.
In most cases, it is indicated following subtotal tumor resection.
In cases of tumor dissemination, craniospinal irradiation is recommended.
Advanced radiation techniques such as intensity-modulated radiation therapy (IMRT) and proton therapy help minimize damage to healthy tissues.
Chemotherapy
Chemotherapy exhibits limited efficacy and is primarily used in children under 1–1.5 years of age to delay radiotherapy or in cases of recurrence where surgery or irradiation is not feasible. Agents used include cisplatin, carboplatin, cyclophosphamide, etoposide, and methotrexate.
In children over 1 year of age and in adults, the role of chemotherapy is restricted due to the low sensitivity of ependymomas to most cytotoxic agents. In rare cases, chemotherapy may be considered for recurrent disease or when surgical and radiation options are unavailable.
Targeted Therapy and Immunotherapy
Experimental data suggest potential for targeted therapy based on molecular profiling (e.g., EGFR and VEGF inhibitors). However, clinical application remains limited and requires further investigation.
Find more scientifically accurate content on our social media
Subscribe and don’t miss out the latest resources
Prognosis and Follow-Up
Survival rates depend on patient age, extent of resection, molecular tumor subtype, and presence of dissemination.
In pediatric patients, five-year survival exceeds 70 % following complete resection and appropriate therapy.
In adults, prognosis also varies by tumor type but is generally favorable following total resection and radiotherapy.
A long-term follow-up period is essential, with regular MRI scans for at least five years to detect late recurrences.
FAQ
1. What is an ependymoma and where can it be found?
An ependymoma is a glial CNS tumor that originates from cells lining the ventricles of the brain and the central canal of the spinal cord. In children, up to 90 % of ependymomas are located in the brain, predominantly in the posterior fossa. In adults, approximately 65 % of cases occur in the spinal cord.
2. What are the characteristic symptoms of ependymoma?
Ependymoma symptoms vary by tumor location: Intracranial ependymomas may present with signs of increased intracranial pressure (headache, nausea), seizures, ataxia (impaired coordination), and focal neurologic deficits. Spinal ependymomas, including those in the cauda equina region, are typically associated with back pain, muscle weakness in the lower extremities, and pelvic organ dysfunction (urinary and bowel disturbances).
3. Is ependymoma a malignant or benign tumor?
Ependymomas represent a heterogeneous group of tumors. Some variants, such as subependymoma (WHO Grade I), are slow-growing and benign. However, the majority are classified as Grade II–III and are considered malignant. The current WHO classification emphasizes molecular features, which more accurately define malignancy grade and prognosis.
4. How is ependymoma treated?
The cornerstone of treatment is complete surgical resection of the tumor. As radical removal is not always feasible, postoperative radiotherapy becomes a critical component of disease control. Chemotherapy plays a limited role and is primarily used in young children to delay irradiation or in cases of recurrence when surgery and radiotherapy are not viable options.
5. What is the prognosis for ependymoma and how long do patients live?
Prognosis and survival rates vary widely and depend on the molecular subtype, patient age, and — most importantly — the extent of surgical resection. In pediatric patients, five-year survival exceeds 70 % following complete resection and subsequent radiotherapy. In adults, prognosis is also generally favorable with adequate treatment. However, aggressive molecular subtypes, such as PF-EPN-A in children or MYCN-amplified spinal ependymoma, are associated with poorer outcomes.
References
1.
VOKA Catalogue. [Electronic resource].
https://catalog.voka.io/
2.
Upadhyaya SA, Tinkle C. Intracranial ependymoma and other ependymal tumors. UpToDate. 2023
3.
Kresbach C, Neyazi S, Schüller U. Updates in the classification of ependymal neoplasms: The 2021 WHO Classification and beyond. Brain Pathol. 2022 Jul;32(4):e13068. doi: 10.1111/bpa.13068. Epub 2022 Mar 21. PMID: 35307892; PMCID: PMC9245931.
4.
Mu W, Dahmoush H. Classification and neuroimaging of ependymal tumors. Front Pediatr. 2023 May 23;11:1181211. doi: 10.3389/fped.2023.1181211. PMID: 37287627; PMCID: PMC10242666.
5.
Mack SC, Witt H, Pajtler KW et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. 2018;553(7686):101-5.
6.
Parker M et al. C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma. Nature. 2014;506(7489):451-5.
7.
Yamaguchi J et al. Latest classification of ependymoma in the molecular era and advances in its treatment: a review. Jpn J Clin Oncol. 2023;53(8):653-663.
8.
Merchant TE et al. Conformal radiotherapy after surgery for paediatric ependymoma: overall survival and toxicity. Lancet Oncol. 2009;10(3):258-66.
9.
National Comprehensive Cancer Network (NCCN) CNS Cancers Guidelines, 2024.
10.
Tsang DS et al. Reirradiation for recurrent pediatric intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2018;100(2):507-514.