The content on this website, including text, graphics, and other materials, is provided for informational purposes only. It is not intended as advice or guidance. Regarding your specific medical condition or treatment, please consult your healthcare provider.
Hypertrophic cardiomyopathy (HCM) is a primary myocardial disorder marked by unexplained, most often asymmetric, hypertrophy of the left ventricular (LV) wall without chamber dilation. The disease is caused by mutations in genes encoding sarcomeric proteins, leading to morphological, electrical, and hemodynamic abnormalities.
HCM is one of the most common inherited forms of cardiomyopathy, occurring in approximately 1 in 500 adults. Men are 1.5 times more likely to develop HCM than women; in women, however, the disease is usually diagnosed later in life and tends to be more severe.
Etiology
HCM results from impaired synthesis and function of sarcomeric contractile proteins. In some cases, secondary forms are identified, associated with systemic or metabolic disorders.
Furthermore, up to 60–70 % of HCM cases are linked to single-gene mutations.
Genes Associated with HCM
Gene
Protein
Mutation Frequency
MYH7
β-myosin heavy chain
MYH7 and MYBPC3 account for 70 % of mutations
MYBPC3
Myosin-binding protein C
MYH7 and MYBPC3 account for 70 % of mutations
TNNT2
Troponin T
Approximately 5 %
TNNI3
Troponin I
< 5 %
TPM1
Tropomyosin
< 5 %
The disease is inherited in an autosomal dominant pattern, meaning that a single altered gene from one parent is sufficient to cause the condition. Thus, the likelihood of developing the disease is high, but the severity and clinical manifestations may vary greatly, even within the same family.
It should be noted that in approximately 30 % of cases, the mutation occurs de novo, without a family history.
Certain disorders may mimic the clinical features of HCM but arise from different pathogenetic mechanisms. It is critically important to distinguish these conditions from the primary (sarcomeric) form, since both treatment strategies and prognosis differ.
Forms with Secondary Hypertrophy (HCM Phenocopies)
Disease
Mechanism
Clinical Features
Fabry disease
Inherited lysosomal storage disorder (α-galactosidase A deficiency)
An autosomal recessive disease associated with FXN gene mutation (mitochondrial disease)
Progressive neurodegeneration: ataxia, muscle weakness and atrophy, speech impairment, etc.
Glycogen storage diseases (e.g., Pompe disease)
Glycogen accumulation in lysosomes, especially in muscle and heart
Skeletal muscles are often involved
Systemic arterial hypertension
Reactive myocardial hypertrophy
Usually symmetric, with history of hypertensive manifestations
Even in cases of a mutation in a sarcomeric protein gene, the clinical manifestations and course of HCM depend on additional factors, including:
Epigenetic regulators;
Concomitant hypertension;
High-intensity physical activity (especially during adolescence);
Sex and hormonal status (women more often develop obstructive forms, but with later onset);
Family history of sudden cardiac death (SCD).
Pathogenesis
Sarcomeric dysfunction
Mutations in the relevant genes (see Etiology) lead to:
Increased calcium sensitivity;
Reduced contractile efficiency;
Elevated energy demand.
Consequence: Compensatory myocardial hypertrophy develops, predominantly involving the interventricular septum, especially in the region of the left ventricular outflow tract (LVOT).
Hypertrophy and impaired relaxation (diastolic dysfunction)
Thickening of the ventricular walls leads to:
Reduced LV compliance;
Impaired diastolic filling;
Elevated diastolic pressure.
Consequence: Pulmonary congestion, manifested by dyspnea and other symptoms of heart failure with preserved ejection fraction (HFpEF).
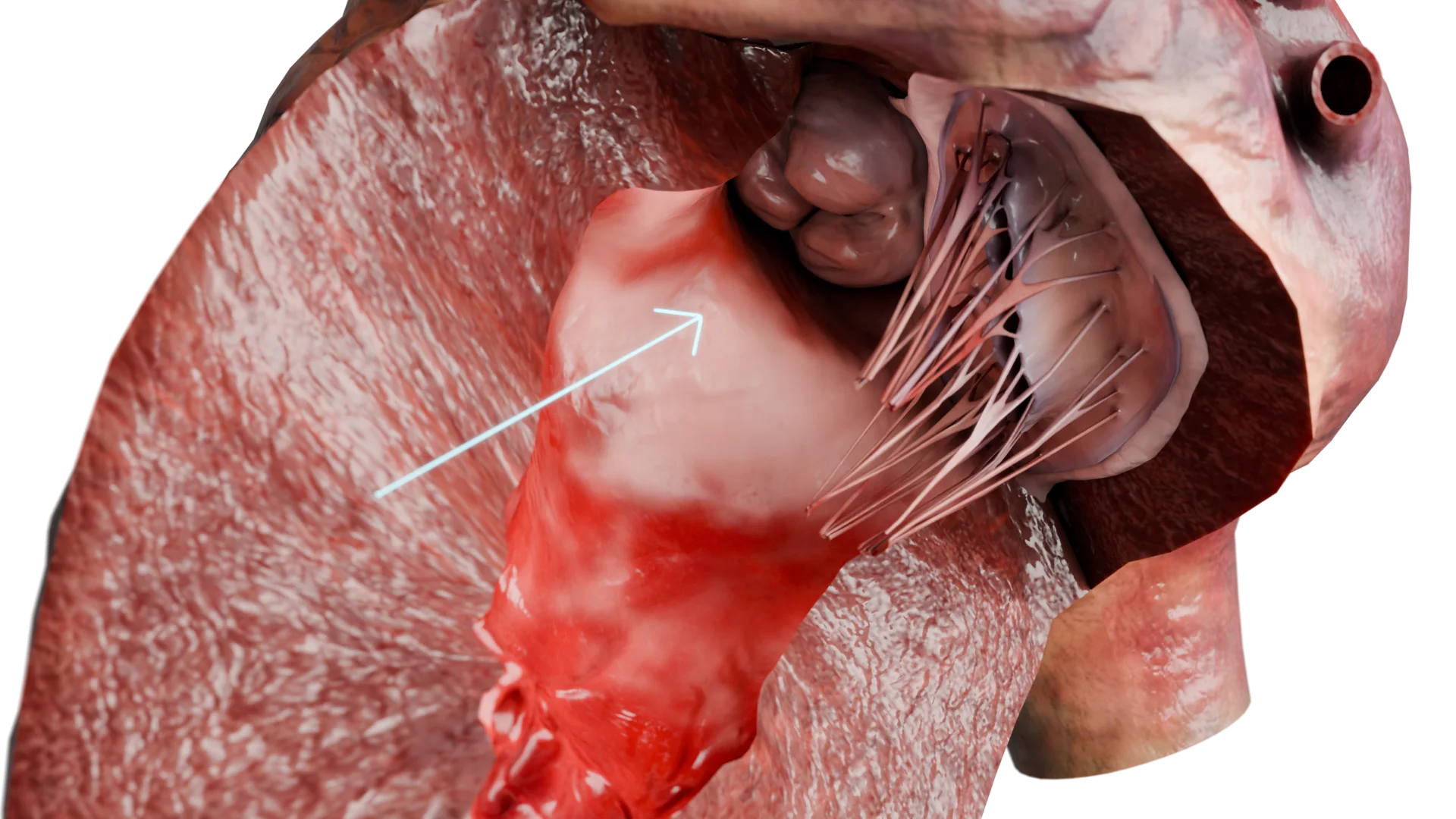
Significant narrowing of LVOT (obstructive form)
LVOT obstruction develops in 60–70 % of patients.
Due to the Venturi effect during systole, the anterior leaflet of the mitral valve is drawn toward the interventricular septum (systolic anterior motion, SAM phenomenon). This increases the pressure gradient and leads to mitral regurgitation (MR).
Consequence: Progressive hemodynamic overload, which worsens symptoms and increases the risk of arrhythmias.
LVOT Obstruction due to Interventricular Septum Thickening — 3D Model
Microvascular ischemia
The hypertrophied myocardium requires increased oxygen supply, but:
The capillary network cannot adequately compensate for tissue growth;
Oxygen demand and supply are out of balance;
Fibrosis of small-caliber vessels is frequently observed.
Consequence: Myocardial ischemia occurs despite normal coronary arteries, leading to chest pain, fibrosis, and an increased risk of arrhythmias.
Fibrosis and electrical instability
In response to ischemia and mechanical overload of the myocardium, interstitial and focal fibrosis develops, resulting in:
Impaired impulse conduction;
Ventricular arrhythmias;
Increased risk of sudden cardiovascular death (SCD).
3D Animation: Hypertrophic Cardiomyopathy
Clinical manifestations
Dyspnea on exertion, fatigue;
Chest pain (angina pectoris) without coronary artery disease;
Syncope or presyncope (especially during physical exertion);
SCD — particularly in younger patients and athletes with obstructive forms;
Heart failure: The condition may develop when ejection fraction (HFpEF) is preserved or reduced (in later stages). Symptoms include edema, orthopnea, tachycardia, and reduced exercise tolerance;
Asymptomatic course: in 25–30 % of patients, the disease is detected during family screening. A high risk of complications is not excluded.
Diagnosis of Hypertrophic Cardiomyopathy
Echocardiography (transthoracic echocardiography, TTE) — the key method for initial diagnosis. It allows:
Assessment of myocardial wall thickness. Diagnosis is likely with wall thickness ≥ 15 mm in adults or ≥ 13 mm in first-degree relatives of patients with confirmed HCM;
Determination of hypertrophy distribution: asymmetric, concentric, apical;
Detection of LVOT obstruction (gradient ≥ 30 mmHg; clinically significant at ≥ 50 mmHg);
Identification of the SAM phenomenon (systolic anterior motion of the mitral leaflet) and MR;
Evaluation of LV function and diastolic dysfunction;
Measurement of atrial size (particularly the left atrium, due to AF risk).
Cardiac MRI is recommended when:
Echocardiography does not allow precise assessment of wall thickness;
Apical or atypical forms of HCM are suspected;
Myocardial fibrosis evaluation is required.
Cardiac MRI identifies:
The distribution and degree of hypertrophy;
Areas of fibrosis associated with increased risk of arrhythmias and SCD;
Differentiation from phenocopies (e.g., amyloidosis).
ECG (echocardiogram) is nonspecific, but pathological changes are detected in more than 90 % of patients. Signs of LVH include:
Atypical Q waves (in leads V4–V6, I, aVL) may mimic myocardial infarction;
ST segment changes and inverted T wave;
Rhythm disturbances: AF, ventricular extrasystoles, ventricular tachycardia (VT);
Verapamil — for patients with contraindications to β-blockers;
Disopyramide — as an adjunct in obstructive forms;
Mavacamten — a novel agent with proven efficacy in reducing LVOT pressure gradient and improving symptoms (ESC 2023, AHA 2020).
Surgical therapy
Indications:
LVOT pressure gradient ≥ 50 mmHg at rest or following provocation;
Severe symptoms (NYHA Class III–IV) unresponsive to β-blockers, verapamil, or disopyramide;
Significant MR associated with SAM phenomenon;
Extended Septal Myectomy:
This is the gold standard surgical treatment for obstructive HCM;
The procedure is performed via mini-sternotomy or full sternotomy; in some cases, via right anterior mini-thoracotomy;
Resection of hypertrophied interventricular septal tissue eliminates the pressure gradient;
Mitral valve repair or resection of secondary chordae may be performed if necessary;
Possible complications: conduction blocks, need for ICD, recurrence of gradient.
Alcohol septal ablation (endovascular intervention) is more often used in patients with contraindications to open surgery or high surgical risk.
Injection of ethanol into a perforating artery → localized infarction → thinning of the septum;
Risk of complete atrioventricular block (up to 10 %) may necessitate pacemaker implantation;
Less predictable outcomes;
Possibility of incomplete relief of obstruction.
Implantable Cardioverter-Defibrillator (ICD)
Indications:
Prior SCD or sustained VT;
LV wall thickness > 30 mm;
Family history of SCD;
Unexplained syncope;
LV ejection fraction < 50 % with progressive disease.
Heart transplantation may be considered in patients with end-stage, refractory heart failure despite optimal therapy.
FAQ
1. What is HCM?
HCM is a condition in which the heart muscle (most often the interventricular septum) becomes abnormally thickened. Unlike hypertrophy due to hypertension or valvular disease, HCM is caused by genetic mutations rather than pressure overload.
2. Is HCM hereditary? Should relatives be screened?
Yes. In most cases, HCM is inherited in an autosomal dominant pattern. Therefore, screening of first-degree relatives is recommended.
3. What symptoms may suggest HCM, and when should you seek medical advice?
Symptoms include shortness of breath, chest pain, dizziness, and fainting, especially during exertion. A cardiologist should be consulted if such complaints occur, or if there is a family history of SCD.
4. Is HCM dangerous? Can patients live long with it?
If diagnosed in a timely manner and treated properly, most patients live full lives. However, HCM increases the risk of arrhythmias and SCD, particularly without treatment.
5. What is the difference between obstructive and nonobstructive HCM?
In obstructive HCM, the thickened septum impedes blood flow from the LV, leading to more pronounced symptoms. In nonobstructive HCM, outflow is not impaired.
6. Can patients with HCM participate in sports?
Intense and competitive sports are not recommended. Moderate physical activity may be allowed, but only under medical supervision.
7. What tests are needed to diagnose HCM?
Typical evaluations include ECG, echocardiography, cardiac MRI, 24-hour Holter monitoring, and genetic testing (when indicated).
8. How is HCM treated? Does it require surgery or may it be managed conservatively?
Pharmacological therapy is the cornerstone of treatment. In severe cases, surgery or percutaneous septal ablation may be required.
9. What is an ICD, and when is it used in HCM?
An ICD (implantable cardioverter-defibrillator) is a device that prevents sudden death from arrhythmias. It is implanted in high-risk patients, based on clinical recommendations.
10. Can HCM be completely cured? What is the prognosis?
HCM cannot be fully cured, but symptoms can be effectively controlled. With proper management, prognosis is generally favorable, especially when no severe complications develop.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.