The content on this website, including text, graphics, and other materials, is provided for informational purposes only. It is not intended as advice or guidance. Regarding your specific medical condition or treatment, please consult your healthcare provider.
Dilated cardiomyopathy (DCM) is a myocardial disease characterized by dilation (enlargement) and systolic dysfunction of the left or both ventricles in the absence of coronary heart disease, congenital malformations, hypertension, and valve abnormalities that could explain these changes. According to population-based studies, the prevalence of DCM is about 0.036–0.400%.
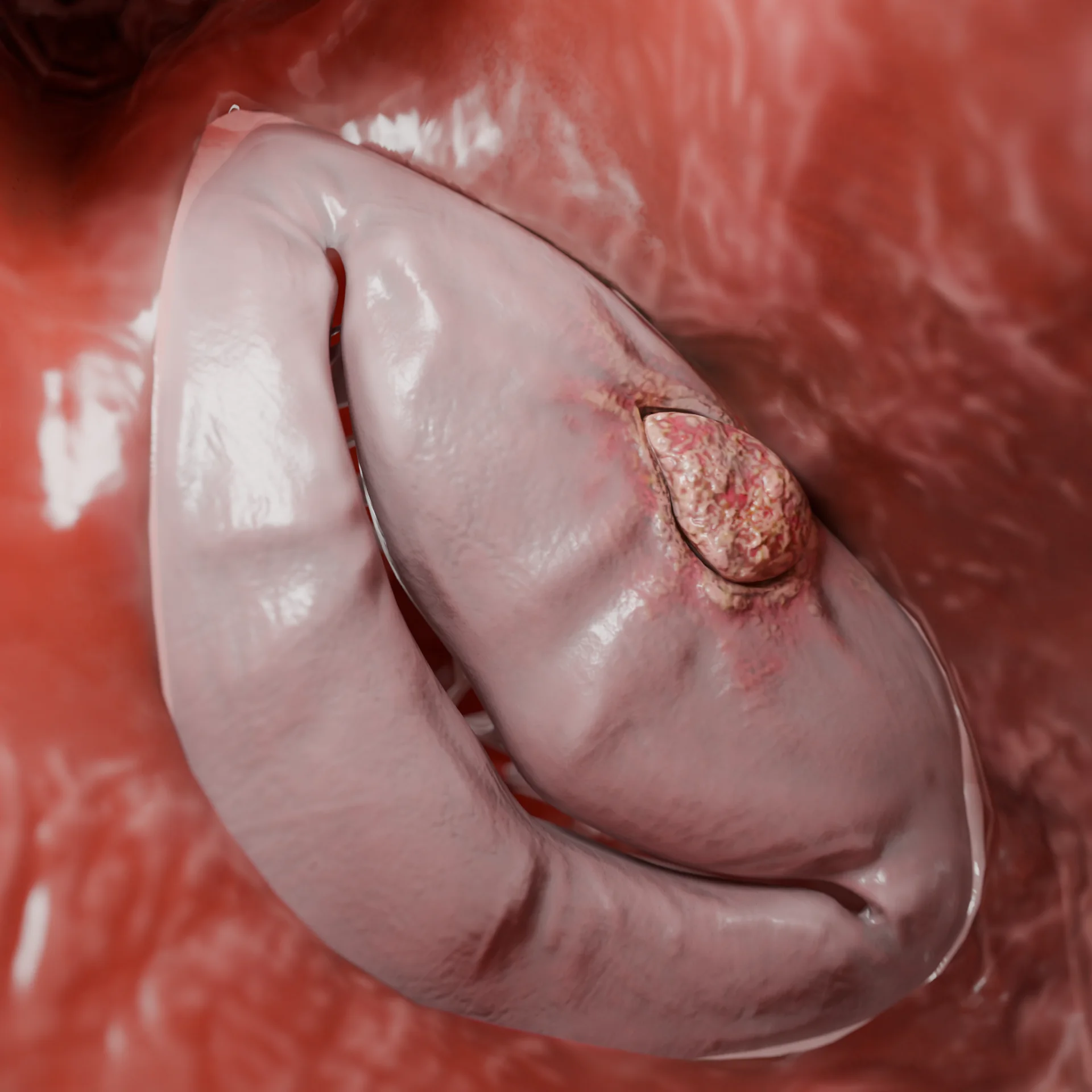
3D Animation: dilated cardiomyopathy
Etiology
The etiology of dilated cardiomyopathy (DCM) is very heterogeneous and includes inherited (genetic/family) and acquired causes:
Genetic causes
It is inherited in a predominantly autosomal dominant manner.
Up to 50% of cases may be familial in nature.
The major genes are TTN, LMNA, FLNC, BAG3, DSP, RBM20, MYH7, SCN5A.
May be combined with arrhythmias, conduction abnormalities, or an overlap phenotype (e.g., with evidence of arrhythmogenic CM).
Inflammatory (post-myocarditis) causes
Often secondary to viral infection (parvovirus B19, HHV-6, adenoviruses, enteroviruses).
Can be autoimmune, associated with diseases such as systemic lupus erythematosus, sarcoidosis, rheumatoid arthritis, etc.
Toxic exposures
Alcohol has a direct myocardial toxicity, especially at high doses and with prolonged use.
Diseases of accumulation: hemochromatosis, Fabry disease, amyloidosis.
Tachyarrhythmias
Prolonged untreated atrial fibrillation, atrial or ventricular tachycardia, paroxysmal tachycardia.
May be reversible with HR/rhythm control.
Peripartum cardiomyopathy
Occurs in the last months of pregnancy or within 5 months of delivery.
Idiopathic cases
A diagnosis of exclusion when no secondary causes are identified and genetic testing is uninformative.
Pathogenesis of dilated cardiomyopathy
Regardless of the cause, the pathogenetic mechanisms are similar: cardiomyocyte damage, inflammation activation, myocardial remodeling and progressive deterioration of contractile function.
Stages of dilated cardiomyopathy:
Primary damage to cardiomyocytes leading to activation of inflammation:
Genetic: disruptions in structure for sarcomere, nucleus, cytoskeleton, etc.;
Toxic: accumulation of free radicals, mitochondrial dysfunction, direct damage to cardiomyocyte membranes;
Viral: chronic inflammation and fibrosis as a result of autoimmune myocarditis.
Disorder of intracellular calcium metabolism, energy metabolism and cardiomyocyte apoptosis.
Myocardial remodeling: wall thinning, cavity dilation (mainly of the left ventricle, less often of both ventricles), development of interstitial fibrosis. As a consequence of annular dilation and papillary muscle dysfunction, atrioventricular valve insufficiency often develops.
The compensatory activation of neurohormonal systems (RAAS, sympathoadrenal), which improves hemodynamics in the short term, can lead to congestive heart failure over time.
Thin left ventricular wall and cavity dilation: 3D modelMitral insufficiency due to dilation of LV cavity and valve annulus: 3D model
Clinical manifestations
Symptoms of dilated cardiomyopathy are due to progressive systolic dysfunction and stasis both in the systemic and pulmonary circulation:
Dyspnoea on exertion, eventually changing to dyspnoea at rest;
Fatigue, decreased tolerance to physical activity;
Orthopnoea and paroxysmal nocturnal dyspnoea;
Swelling of the lower extremities;
Liver enlargement, ascites;
Syncope, dizziness (possible with arrhythmias or low blood pressure);
Tachycardia, rhythm disorders (especially atrial fibrillation and ventricular arrhythmias);
Less commonly, chest pain due to subendocardial ischaemia.
Diagnosis of dilated cardiomyopathy
The diagnosis of DCM is based on the finding of left ventricular dilation and systolic dysfunction not explained by ischaemia, hypertension, valve malformations, or congenital pathology. The purpose of diagnosis is to confirm the cardiomyopathic phenotype, determine the causes and assess the severity of changes in cardiac structures, as well as the likelihood of developing adverse effects.
Instrumental methods
Cardiac US is the main method of primary diagnosis:
LVEDV is increased (>150–180 mL or indexed >75 mL/m²);
Reduced ejection fraction (<45%);
Global hypokinesis without regional abnormalities;
Often: mitral and tricuspid regurgitation, pulmonary hypertension, dilation of RV.
Cardiac MRI with gadolinium (late gadolinium contrast):
Clarifies myocardial structure and tissue characteristics: fibrosis, edema, fatty infiltration;
Typical pattern in DCM: meso- or subepicardial accumulation of gadolinium in the lateral or septal wall;
Indispensable in suspected myocarditis, sarcoidosis, and accumulation diseases.
Is performed to rule out CHD in patients over 35 years of age or in the presence of risk factors;
Mandatory for typical chest pain, regional abnormalities on cardiac US or late gadolinium accumulation.
Holter monitor or ambulatory ECG (24–72 h):
Ventricular arrhythmias (VE, VT), AF, tachycardia, pause, block (especially if LMNA-mutant form is suspected);
Assists in selecting therapies and deciding on an ICD.
Myocardial biopsy (as indicated):
If suspected: active myocarditis, infiltrative diseases (amyloidosis, sarcoidosis);
Limited use, requires accurate readings and a high level of execution.
Laboratory Methods
BNP/NT-proBNP:
It is the most sensitive biomarker of congestive heart failure. Levels rise in proportion to the degree of volume and pressure overload. High values indicate decompensation, low values allow to exclude HF.
Cardiac-specific troponins (I or T):
Moderate elevation is possible with active inflammation (e.g., myocarditis) or marked myocardial distension. Significant and acute elevation requires exclusion of myocardial infarction.
Thyroid hormones (TTG, free T3 and T4):
Hypothyroidism can cause systolic dysfunction, hyperthyroidism can cause tachycardia-induced DCM.
Glucose and glycated hemoglobin (HbA1c):
Diabetes mellitus is associated with the development of diabetic cardiomyopathy and also exacerbates the course of HF.
Allow detection of iron deficiency or hemochromatosis. The latter may lead to secondary cardiomyopathy with progressive LV dysfunction.
Markers of inflammation and autoimmunity (antinuclear antibodies, rheumatoid factor, anticardiolipin antibodies, etc.):
Performed when autoimmune or systemic inflammatory nature of the disease is suspected—systemic lupus erythematosus, scleroderma, myocarditis, etc.
Angiotensin-converting enzyme, soluble interleukin-2 receptor, and calcium:
Used when cardiac sarcoidosis is suspected. Particularly relevant when combined with conductive abnormalities or unclear infiltrative changes on MRI.
Liver tests, creatinine, electrolytes:
Tested routinely to determine systemic manifestations of heart failure and to assess tolerability of therapy.
Genetic testing:
Indicated for family history of cardiomyopathy, sudden cardiac death, blocks, severe dysfunction at a young age or no secondary causes. Panels for genes associated with DCM (most commonly TTN, LMNA, BAG3, BAG3, FLNC, SCN5A, etc.) are used.
Find more scientifically accurate content on our social media
Subscribe and don’t miss out the latest resources
Treatment of dilated cardiomyopathy
Medical Therapy
Drug therapyfor DCM requires a comprehensive and strictly individualized approach, considering the clinical and functional characteristics of the patient.
SGLT2 inhibitors (dapa/empagliflozin): improved prognosis regardless of the presence of diabetes;
Diuretics: for symptoms of fluid retention;
Ivabradine: when HR >70 in sinus rhythm if beta-blockers are inadequate;
Anticoagulants: in case of AF, presence of blood clots, high HR.
Surgical Therapy
Device implantation:
ICD (cardioverter-defibrillator): if EF <35%, NYHA II-III and risk of VT;
CRT-P/CRT-D (resynchronization therapy): if QRS >130 ms, EF <35%, sinus rhythm.
Surgical correction of mitral regurgitation (secondary) in case of:
Functional mitral regurgitation of grade II to III;
LVEF 30–50%, LV DCD <70 mm (no significant LV dilation);
Presence of HF symptoms despite drug therapy;
Techniques: annuloplasty (reduction of the annulus fibrosus), flap reconstruction, in some cases MitraClip (via catheter correction).
Mechanical circulatory support (LVAD: HeartMate, HeartWare): Indications:
Severe HF (NYHA IV) refractory to treatment;
Candidates for heart transplantation (as a bridge-to-heart-transplantation);
Patients who are not eligible for transplantation (as long-term supportive care therapy).
Heart transplantation:
Severe refractory HF (NYHA IIIb-IV);
Ineffectiveness of medication and device therapy;
Progressive decline in target organ function;
Age usually <65 years, no absolute contraindications;
Contraindications: malignant tumors (with poor prognosis of life expectancy), active infections, severe pulmonary hypertension, compliance disorders.
FAQ
1. Can dilated cardiomyopathy be completely cured?
Not completely, but with proper treatment it is possible to significantly improve quality and length of life.
2. What is the difference between idiopathic and genetic forms of DCM?
Idiopathic has no identified cause; genetic is caused by inherited mutations.
3. Why can DCM develop in people without heart disease?
It can be caused by hidden genes, viruses, toxins, hormonal disorders or overload.
4. How life-threatening is DCM and what are its major complications?
Dilated cardiomyopathy is a life-threatening disease that progresses without treatment, but timely therapy can significantly reduce the risks. Its danger stems from the development of three key complications: progressive heart failure, which leads to multiorgan dysfunction; malignant ventricular arrhythmias, causing sudden cardiac death; and thromboembolic events, which can lead to fatal stroke.
Yes, up to 50% of cases have a familial form. ECG and cardiac US are recommended for relatives.
7. What does “reduced ejection fraction” mean?
This is an indicator of the pumping function of the heart. In DCM, it is reduced due to weakening of the cardiac muscle.
8. When should a defibrillator (ICD) be placed in DCM?
In severe reduction of EF (<35%) and risk of severe arrhythmias.
9. Can you exercise with DCM?
Only moderate exercise, agreed with a cardiologist, is allowed.
10. Can you get pregnant with DCM?
It is possible, but in stable condition and under strict medical supervision — the risks depend on the severity of the disease.
11. What are the features of dilated cardiomyopathy in children?
In children, DCM is more often associated with past myocarditis or specific genetic syndromes. The clinical presentation may be nonspecific (dyspnoea, feeding difficulties) and the prognosis is often more serious than in adults.
References
1.
VOKA Catalog. [Electronic resource].
https://catalog.voka.io/
2.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi: 10.1093/eurheartj/ehad194.
3.
Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet. 2023 Sep 16;402(10406):998-1011. doi: 10.1016/S0140-6736(23)01241-2.
4.
Gigli M, Stolfo D, Merlo M, et al. Pathophysiology of dilated cardiomyopathy: from mechanisms to precision medicine. Nat Rev Cardiol. 2025 Mar;22(3):183-198. doi: 10.1038/s41569-024-01074-2.
5.
Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature. J Intern Med. 2019 Oct;286(4):362-372. doi: 10.1111/joim.12944.
6.
Peters S, Johnson R, Birch S, et al. Familial Dilated Cardiomyopathy. Heart Lung Circ. 2020 Apr;29(4):566-574. doi: 10.1016/j.hlc.2019.11.018.
7.
Harding D, Chong MHA, Lahoti N, et al. Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy. J Intern Med. 2023 Jan;293(1):23-47. doi: 10.1111/joim.13556.