Plexus-chorioideus-Tumoren: Plexuspapillome und Plexuskarzinome. Ätiologie, Klassifikation und Therapie
Gontsov A.Neuro-onkologischer Chirurg, MD
11 min lesen·Dezember 11, 2025
Dieser Artikel dient nur zu Informationszwecken
Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
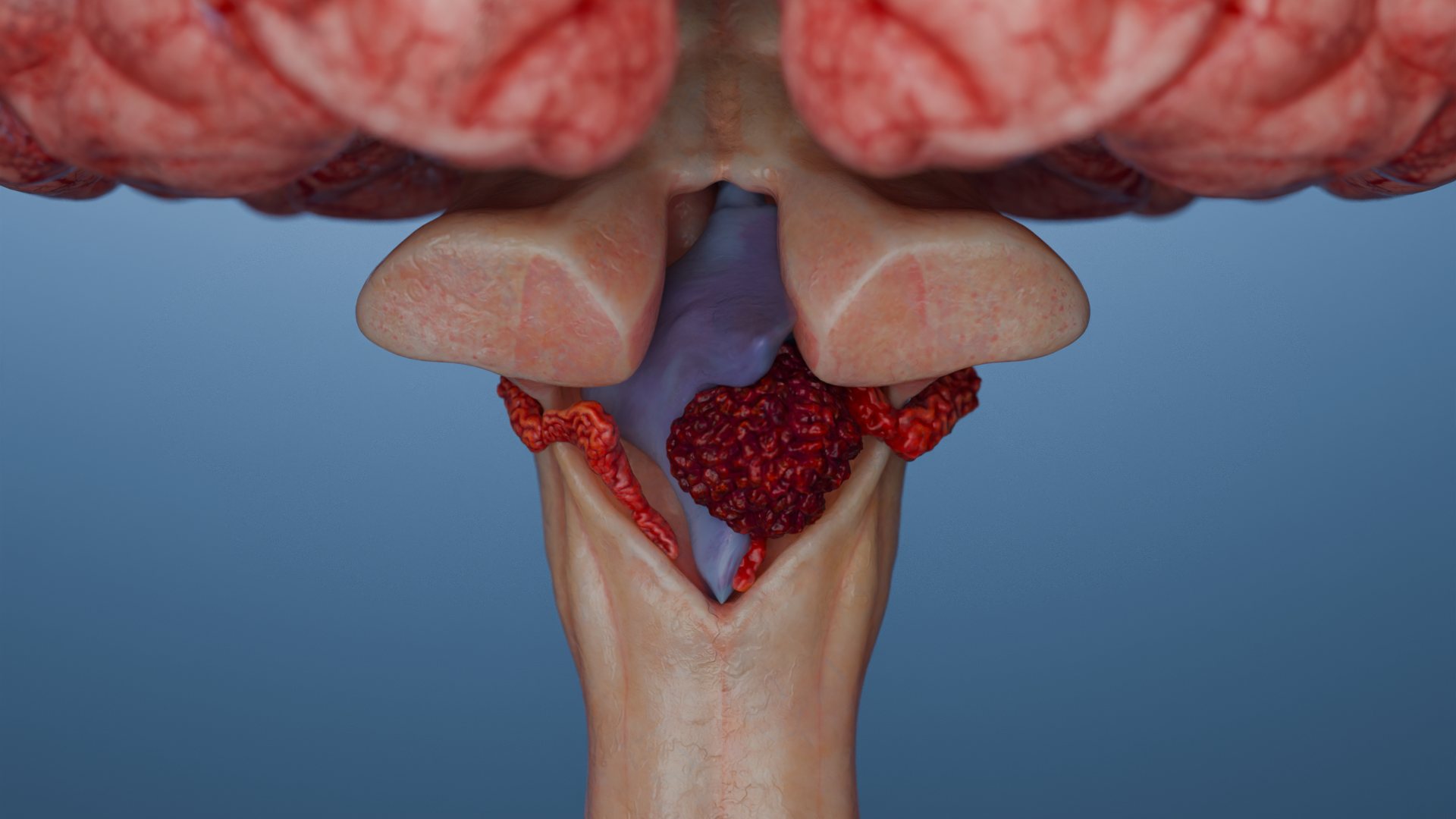
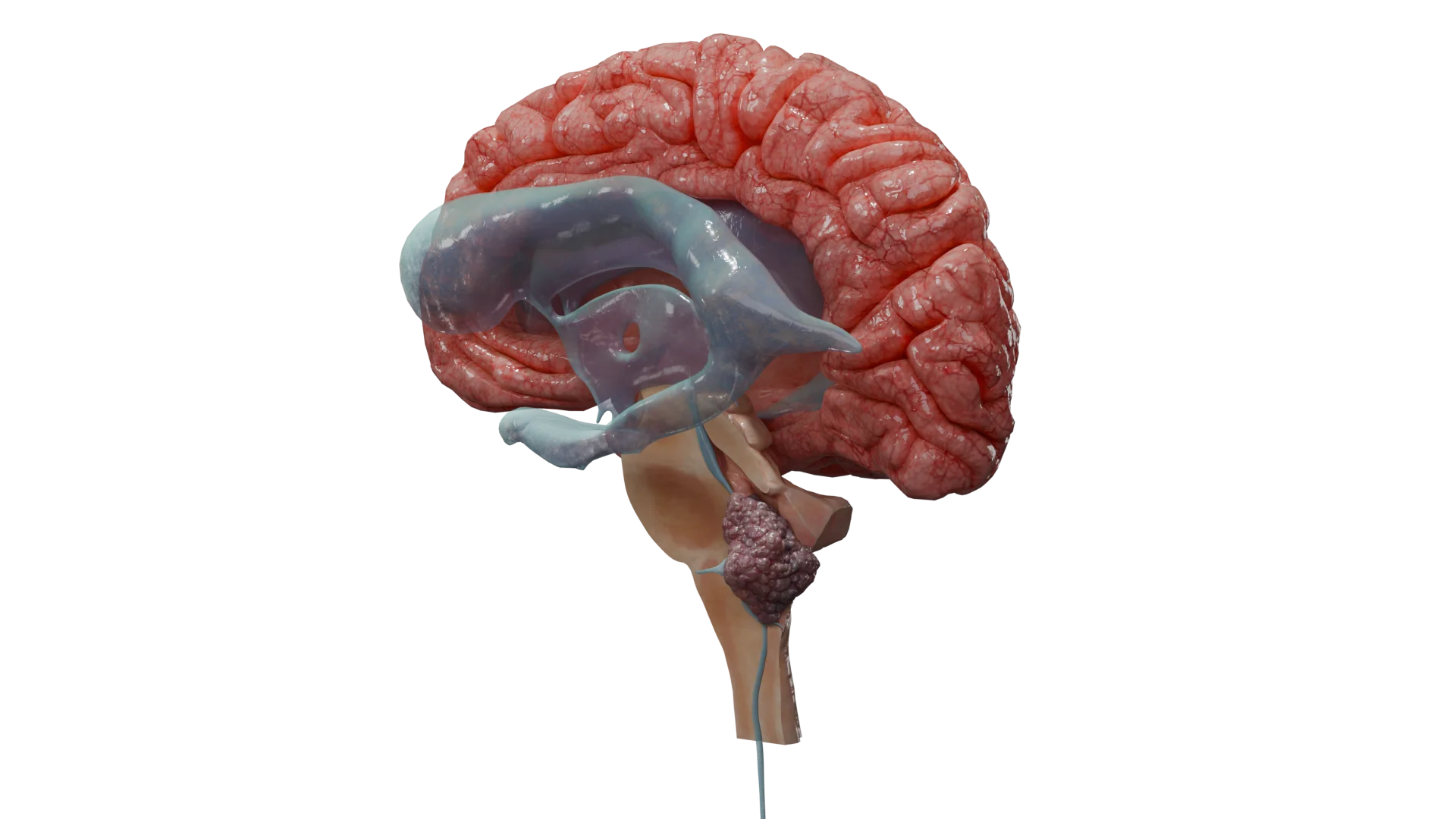
Der Plexus chorioideus produziert Liquor (Liquor cerebrospinalis, Hirnwasser) und befindet sich in den Hirnventrikeln. Die Lokalisation des Tumors hängt vom Alter ab. Bei Kindern sind häufiger die Seitenventrikel betroffen, bei Erwachsenen der IV. Ventrikel.
Plexus-chorioideus-Papillome (Plexuspapillom, Choroidplexuspapillom, CPP) sind benigne Tumoren, die vom Epithel des Plexus chorioideus des Gehirns ausgehen. WHO Grad I. Es ist die häufigste Tumorform des Plexus chorioideus.
Plexus-chorioideus-Karzinome (Plexuskarzinom, Choroidplexuskarzinom, CPC) sind seltene hochmaligne Tumoren (WHO Grad III), die vom Epithel des Plexus chorioideus der Hirnventrikel ausgehen. Ganz überwiegend sind Kinder unter 5 Jahren betroffen. Der Tumor weist einen aggressiven Verlauf auf, neigt zu Invasion und Metastasierung innerhalb des ZNS und hat eine äußerst schlechte Prognose.
Ätiologie und Epidemiologie der Tumoren
Ätiologie und genetische Faktoren
Die genauen Ursachen von Plexuspapillomen sind unklar. Für CPP sind Treibermutationen untypisch, jedoch wird eine Reihe von Fällen mit seltenen genetischen Syndromen in Verbindung gebracht, beispielsweise mit dem Aicardi-Syndrom.
Atypische (aCPP) und bösartige Formen (CPC): Sie können im Zusammenhang mit TP53-Mutationen auftreten, insbesondere bei Kindern mit Li-Fraumeni-Syndrom.
Die Mehrzahl (bis zu 60%) der Patienten mit CPC weisen eine somatische TP53-Mutation auf, und bei 40% steht der Tumor im Zusammenhang mit dem Li-Fraumeni-Syndrom (LFS), das durch eine Keimbahnmutation TP53 verursacht wird.
In einigen Fällen werden andere Störungen in den Genen entdeckt, die den Zellzyklus steuern (z. B. in PTEN, CDKN2A).
Epidemiologie
Die epidemiologischen Merkmale von Plexus-chorioideus-Tumoren basieren auf ihrer geringen Häufigkeit und einer starken Abhängigkeit vom Alter des Patienten.
Häufigkeit: Plexus-chorioideus-Tumoren kommen selten vor (weniger als 1% aller intrakraniellen Tumoren). Bei Kindern unter 1 Jahr machen diese Tumoren 10–15% aller intrakraniellen Neubildungen aus.
Alter: Der Erkrankungsgipfel fällt auf die ersten drei Lebensjahre.
Lokalisation: Bei Kindern sind diese Tumoren häufiger in den Seitenventrikeln und bei Erwachsenen im IV. Ventrikel lokalisiert. Das Geschlechterverhältnis ist etwa 1:1.
Klinik und Symptomatik der Plexus-chorioideus-Tumoren
Das Leitsymptom ist eine Erhöhung des Hirndrucks infolge eines Hydrocephalus (erhöhte Liquorproduktion). Es äußert sich durch Kopfschmerzen, und bei Kleinkindern ist die Makrozephalie das auffälligste Symptom.
In Einzelfällen kann es zu einer intrakraniellen Blutung kommen.
3D-Animation — Plexus-chorioideus-Papillom
Klassifikation der Plexus-chorioideus-Tumoren
Die aktuelle Klassifikation (WHO, 5. Auflage aus dem Jahr 2021) beruht auf molekularbiologischen, immunhistochemischen und histologischen Eigenschaften.
Eine erhöhte mitotische Aktivität ist typisch; erhöhtes Rezidivrisiko
Grad III
Choroid-Plexus-Karzinom (Plexuskarzinom, CPC)
Bösartig, invasiv
Molekulare Klassifikation
Aktuelle Studien unterscheiden drei molekulare Subtypen anhand der Methylierung und Genexpression:
Kindlich supratentoriell, low-risk (CPP/aCPP)
Kindlich supratentoriell, high-risk (CPC)
Adult infratentoriell, low-risk (CPP/aCPP)
So werden heute in der klinischen Praxis die Morphologie, die proliferative Aktivität, molekulare Marker und der TP53-Status berücksichtigt, die die Prognose und den Umfang der Therapie bestimmen.
Diagnostik und Differentialdiagnose
Diagnoseverfahren
Das wichtigste Diagnoseverfahren ist MRT. Ist dies nicht möglich, wird eine Computertomographie oder Ultraschalluntersuchung durchgeführt (im frühen Kindesalter über die Fontanelle).
MRT und CT: Plexus-chorioideus-Tumoren sind in der Regel T2-hyperintensiv, weisen häufig Verkalkungen im CT auf und zeigen eine intensive Kontrastmittelaufnahme.
Für Plexuskarzinome sind eine heterogene Struktur, Nekrosen, Invasion, verstärkte KM-Anreicherung und Anzeichen einer vasogenen Ödembildung um den Tumor herum charakteristisch.
Molekulare Diagnostik: Ein Screening auf LFS (TP53-Keimbahnmutation) ist in allen Fällen von CPC obligat.
Lumbalpunktion: Zum Ausschluss von Tumorzellen im Liquor (bei CPC).
Somit konzentriert sich die Differentialdiagnose bei Kindern auf Ependymome und embryonale Tumoren und bei Erwachsenen auf Meningeome und metastatische Karzinome.
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Therapie und Prognose bei Plexus-chorioideus-Tumoren
Die Therapie ist multimodal und umfasst zunächst einen chirurgischen Eingriff, der die weitere Vorgehensweise und Prognose bestimmt.
1. Chirurgische Behandlung
Goldstandard:totale Resektion. Häufig kommen endoskopische Verfahren zum Einsatz.
Prognose bei Plexuspapillomen: Auch eine subtotale Resektion bei klassischem CPP sorgt in der Regel für eine gute Prognose.
Schwierigkeiten bei Plexuskarzinomen: Eine vollständige Entfernung kann sich durch das infiltrative Wachstum und mögliche intraoperative Blutung schwierig gestalten.
Postoperativ: Aufgrund einer Liquorzirkulationsstörung kann ein Shunt erforderlich sein.
2. Bestrahlung
CPP: In der Regel nach einer Totalextirpation nicht erforderlich.
Anzeigen: Nach Rezidiven, wenn eine ausreichende Resektion nicht möglich ist oder bei atypischen Formen.
CPC: Patienten über 3 Jahre erhalten nach einer Chemotherapie eine kraniospinale Bestrahlung.
Einschränkungen: Bei Säuglingen wird sie aufgrund des Risikos kognitiver Störungen vermieden. Bei Kindern < 3 Jahren und bei Li-Fraumeni-Syndrom wird Strahlentherapie wegen des Risikos von Sekundärtumoren äußerst vorsichtig angewendet.
3. Chemotherapie
Wird eingesetzt: bei Hochrisikopatienten (CPC, aCPP mit Residualtumor, Metastasen).
Therapieschemata: Carboplatin/Etoposid/Vincristin (CarbEV), CycEV. CarbEV zeigt eine höhere Überlebensrate bei CPC.
In Einzelfällen wird bei Li-Fraumeni konsolidierende Hochdosis-Chemotherapie mit autologer Knochenmarktransplantation angewendet.
Prognose und Überlebensfaktoren
Gut: Nach der totalen Resektion von CPP/aCPP beträgt die 10-Jahres-Überlebensrate über 90%. Bei aCPP ist das Rezidivrisiko höher, sodass häufiger eine adjuvante (postoperative) Therapie erforderlich ist.
Schlecht: Die Prognose bei Plexuskarzinomen ist schlechter (5-Jahres-Überlebensarte max. 40%) unter TP53-Mutationen. Eine noch schlechtere Prognose besteht bei genetischen Syndromen, Metastasen, inkompletter Resektion sowie bei Patienten mit Li-Fraumeni-Syndrom.
FAQ
1. Was ist der Unterschied zwischen einem Plexuspapillom und einem Plexuskarzinom?
Ein Plexuspapillom (CPP) ist gutartig (WHO Grad I) mit einer guten Prognose, wobei Plexuskarzinome (CPC) hochmaligne Tumoren (WHO Grad III) mit aggressivem Verlauf und schlechter Prognose darstellen.
2. Was ist das Leitsymptom von Plexus-Chorioideus-Tumoren?
Am häufigsten findet sich intrakranielle Hypertension infolge einer gestörten Liquorproduktion (Hydrozephalus). Bei Säuglingen ist die Makrozephalie das auffälligste Symptom.
3. Wie ist die Rolle der TP53-Mutation bei der Diagnostik von Plexuskarzinomen?
Die TP53-Mutation ist äußerst wichtig für die Prognose: Sie ist häufig mit dem Li-Fraumeni-Syndrom assoziiert und deutet auf eine ungünstigere Prognose bei Plexuskarzinomen hin.
4. Welche Therapie gilt als „Goldstandard“ für Plexuspapillome?
Bei Plexus-chorioideus-Papillomen ist es die vollständige chirurgische Entfernung des Tumors (Gross Total Resection).
5. Warum ist die Strahlentherapie bei der Behandlung von CPC bei Kleinkindern möglicherweise hinauszuschieben?
Die Bestrahlung wird bei Kindern unter 3 Jahren wegen des hohen Risikos schwerer kognitiver Störungen und der Sekundärtumoren vermieden, insbesondere bei Patienten mit Li-Fraumeni-Syndrom.
Verweise
1.
VOKA-Katalog. [Elektronische Quelle].
https://catalog.voka.io/
2.
Wolff JE, et al. „Final results of the Choroid Plexus Tumor study CPT-SIOP-2000.“ J Neurooncol 2022;156(3):599–613.
3.
Louis DN, et al. „The 2021 WHO Classification of Tumors of the Central Nervous System: a summary.“ Neuro Oncol. 2021;23:1231–1251.
4.
Thomas C, et al. „Molecular genetics and diversity of choroid plexus tumors.“ Neuro-Oncol Adv 2024.
5.
Andour H, et al. „Atypical choroid plexus papilloma: Diagnosis and management.“ SAGE Open Med. 2024; doi:10.1177/2050313X241254000.
6.
Yankelevich M, et al. „Marrow-ablative consolidation chemotherapy and molecular targeted therapy for CPC.“ Neuro-Oncol Adv 2024;6(1):vdae109.
7.
Cornelius A, et al. „Molecular Guided Therapy Provides Sustained Clinical Remission in Pediatric CPC.“ Frontiers Pharmacol. 2017;8:652.
8.
Wolff JE, Sajedi M, Brant R. „Choroid plexus tumours.“ Br J Cancer. 2002 87:1086–91.
9.
Safaee M, et al. „Choroid plexus papillomas: advances in molecular biology and surgical management.“ Neurosurg Focus. 2012;32(2):E6.
10.
Maurizi, E. et al. „Diagnosis of choroid plexus papilloma.“ Curr Probl Cancer. 2024; doi:10.1016/j.cpt.2023.09.005.
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io