Ependymome: Epidemiologie, Klassifikation, Diagnostik, Therapie und Prognose
Dobriyan S.Chirurgischer Onkologe, MD
17 min lesen·November 13, 2025
Dieser Artikel dient nur zu Informationszwecken
Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
Bei Ependymomen handelt es sich um eine Gruppe von Gliomen, die aus den die Hirnhöhlen und den Rückenmarkskanal auskleidenden Ependymzellen entstehen. Diese Tumoren können in verschiedenen Bereichen des zentralen Nervensystems auftreten, am häufigsten jedoch in der hinteren Schädelgrube und im Rückenmark. Der moderne Ansatz bei Ependymomen basiert auf einer maximalen chirurgischen Entfernung des Tumors mit anschließender Strahlentherapie. Anatomische, histologische und molekulargenetische Merkmale von Ependymomen bestimmen die Prognose.
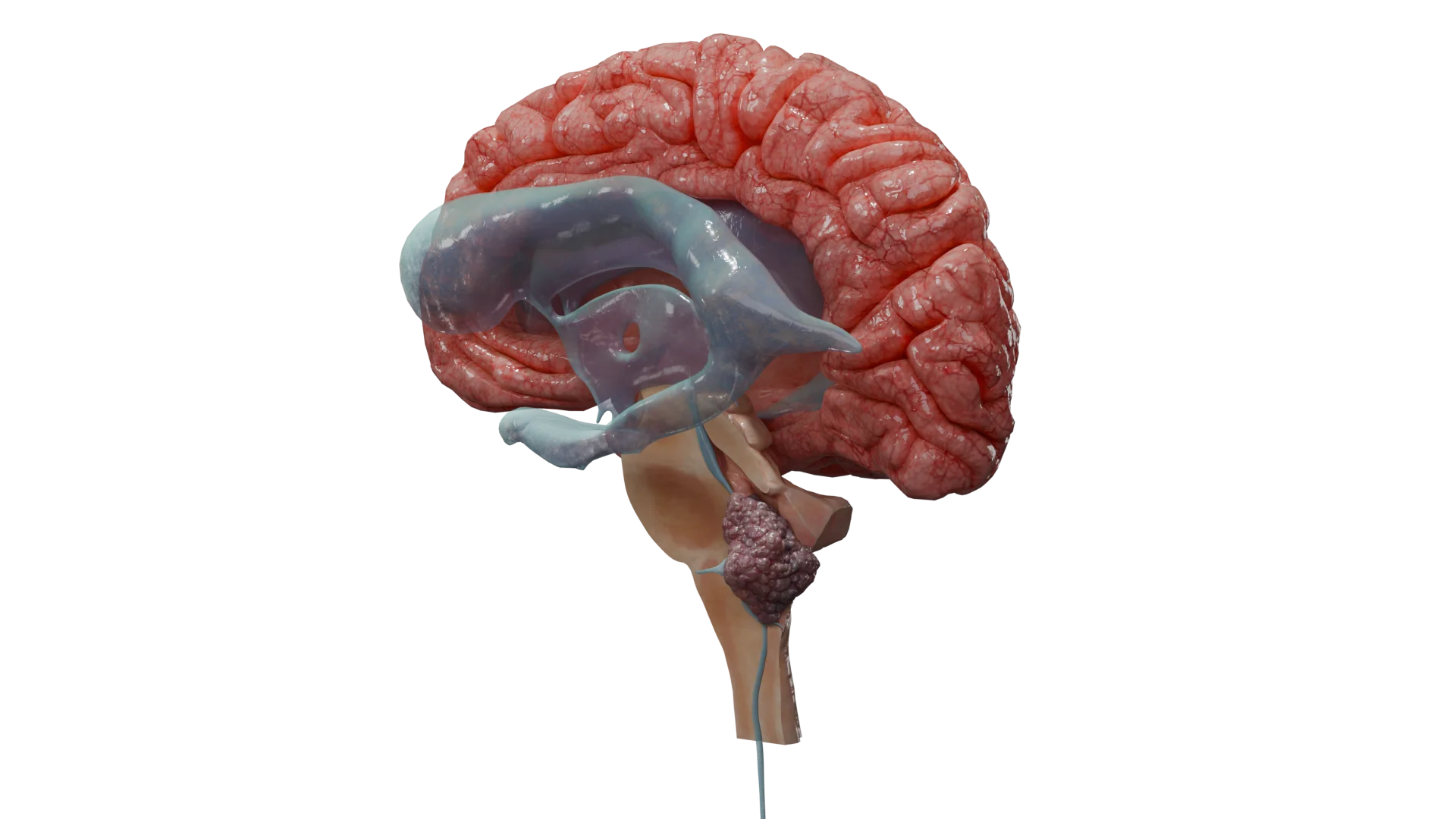
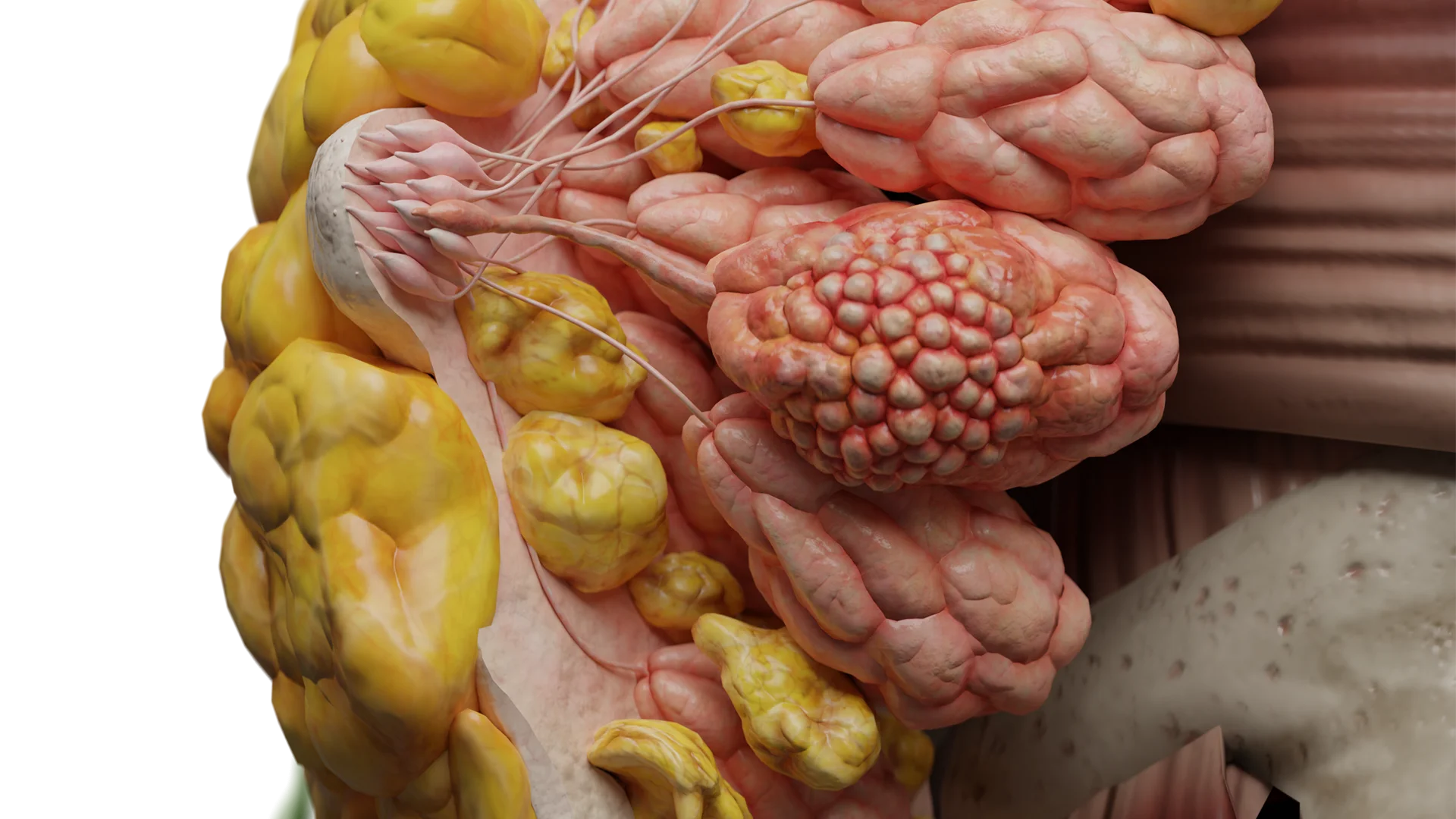
Infratentorielles Ependymom des Gehirns – 3D-Modell
Ätiologie
Genaue Ursachen von Ependymomen sind bisher unklar.
Es werden mehrere Mechanismen unterschieden, die diese Tumoren begünstigen können:
Genetische Veränderungen: Ependymome weisen häufig Veränderungen in der DNA-Struktur auf, u. zw. Ungleichgewicht der Chromosomen (z. B. Chromosomenveränderung +1q), Genfusionen (z. B. ZFTA-RELA-Fusionen), Deletionen und Mutationen, die zur Aktivierung onkogener Signalwege und zur Unterdrückung von Kontrollmechanismen des Zellwachstums führen.
Epigenetische Veränderungen (veränderte Genaktivitäten): von besonderer Bedeutung sind Fehlregulationen der DNA- und Histonmethylierung, u. a. fehlende H3K27me3-Trimethylierung in der PF-EPN-A-Gruppe, wodurch die für die Differenzierung verantwortliche Genexpression unterdrückt wird.
Genetische Faktoren: es besteht ein Zusammenhang zwischen einigen spinalen Ependymomen und NF2-assoziierter Schwannomatose (NF2).
Somit entstehen Ependymome durch eine komplexe Zusammenwirkung von genetischen und epigenetischen Prozessen, die eine Fehlsteuerung des Zellzyklus und abnorme Proliferation bewirken.
Epidemiologie
Ependymome werden in jedem Alter diagnostiziert, wobei die Indizenzspitze im frühen Kindesalter (Median 5 Jahre) liegt.
90% der kindlichen Ependymome sind im Kopf, insbesondere in der hinteren Schädelgrube lokalisiert.
Bei Erwachsenen ist die Inzidenz niedriger. Die meisten Ependymome (65%) finden sich im Rückenmark.
Symptome
Das klinische Erscheinungsbild hängt von der Lokalisation des Tumors ab.
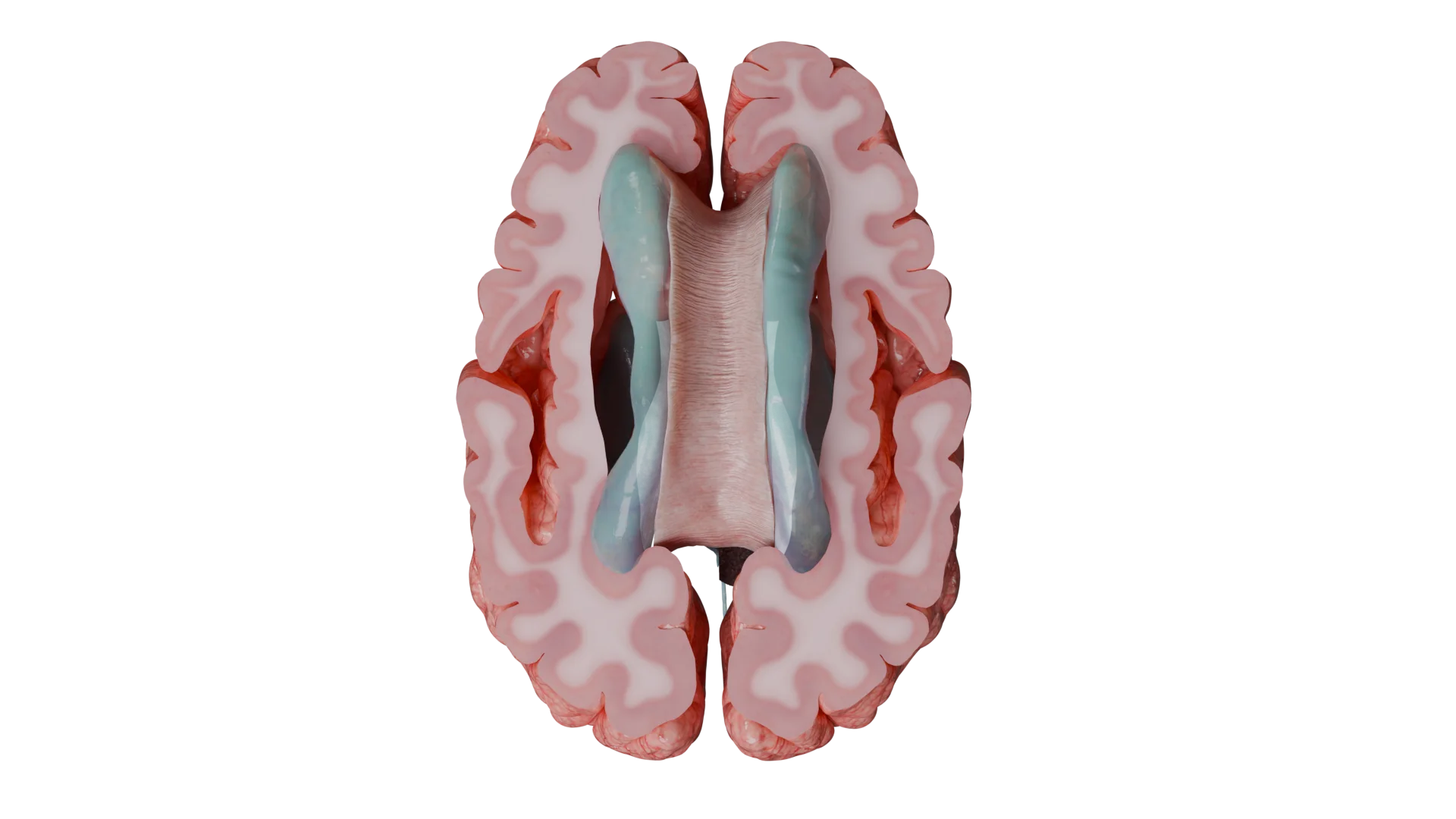
Supratentorielle Ependymome (oberhalb des Tentoriums): Krampfanfälle, fokale neurologische Erscheinungen (Sprachstörungen, Persönlichkeits- und Verhaltensänderungen, Konzentrations- und Gedächtnisschwäche, Hemiparesen), allgemeine zerebrale Symptome (Kopfschmerzen, Übelkeit, Erbrechen, Schwellung der Sehnervenpapille).
Seitenventrikel sind eine typische Lokalisation für supratentorielle Ependymome.
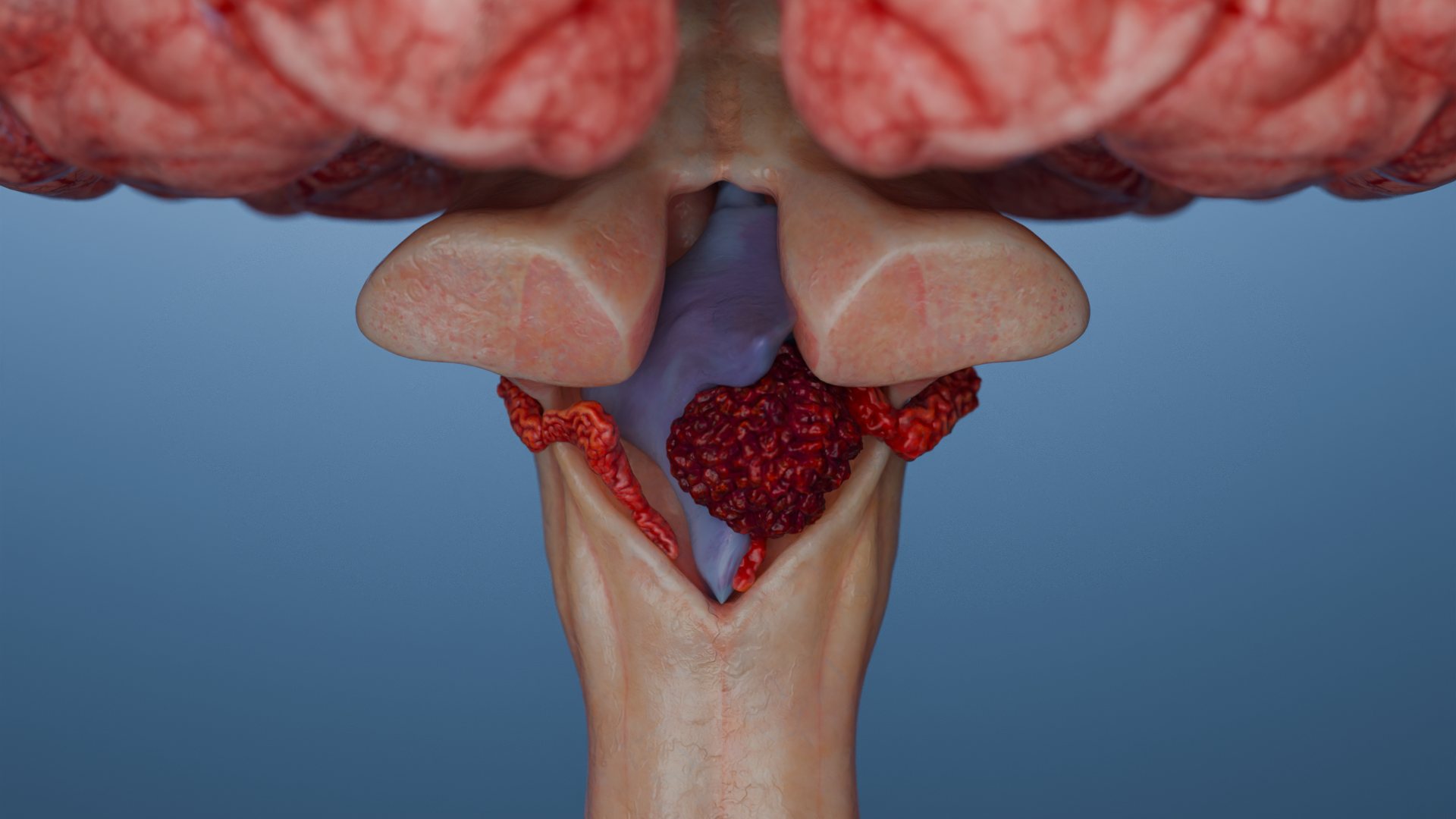
Infratentorielle Ependymome (hintere Schädelgrube): Symptome eines erhöhten Hirndrucks (Hydrozephalus — Kopfschmerzen, Übelkeit, Erbrechen), Ataxie (Koordinationsstörungen und Gangunsicherheit), Funktionsstörungen der Hirnnerven (Dysarthrie, Dysphagie, Hörminderung).
3D-Animation — infratentorielles Ependymom
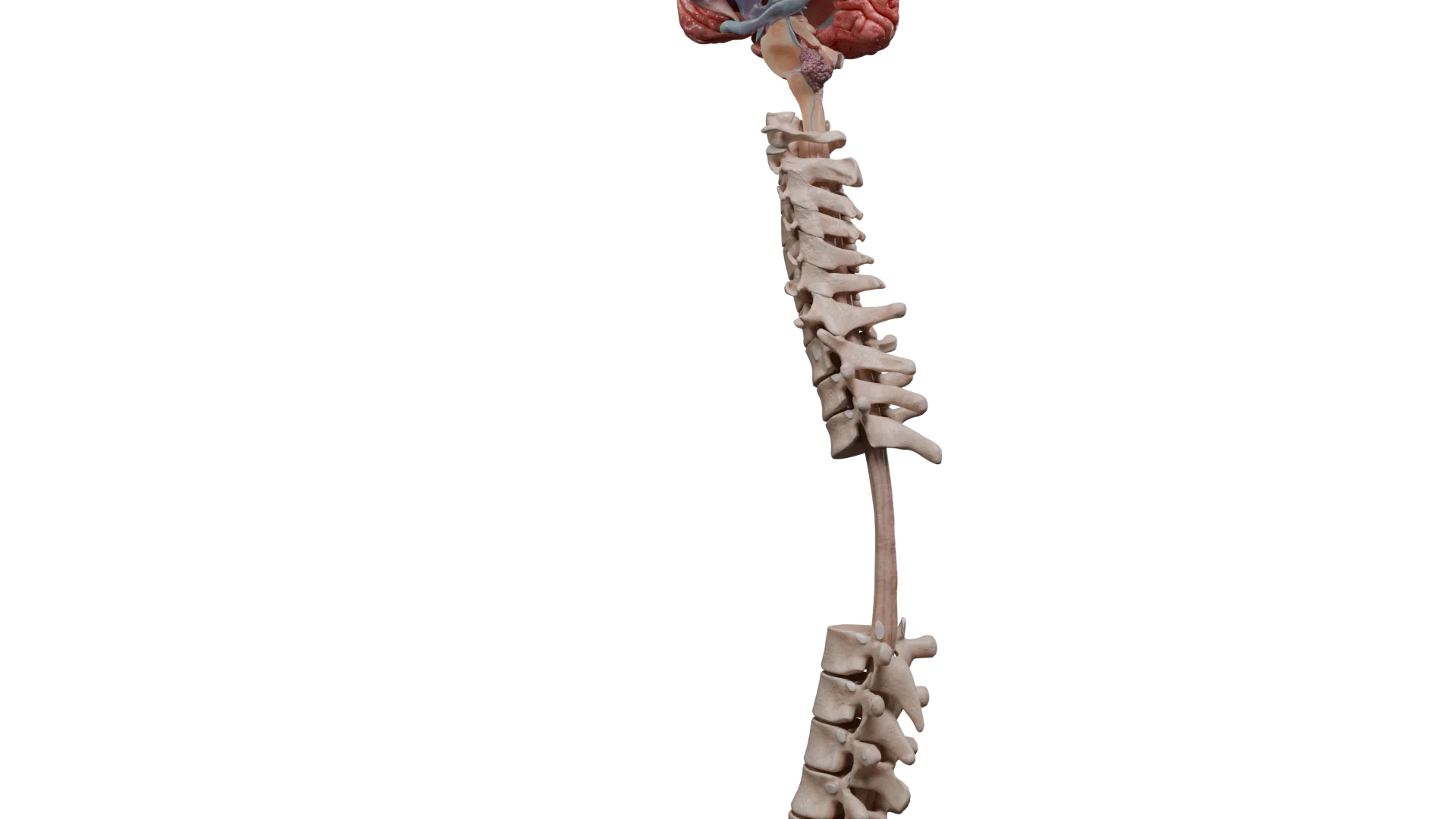
Spinale Ependymome: Rückenschmerzen, Radikulopathie, fortschreitende Muskelschwäche, Funktionsstörungen der Beckenorgane (Miktions- und Defäkationsstörungen, ggf. Impotenz).
Lokalisationen spinaler Ependymome
Klassifikation der Ependymome
Ependymome werden anhand der anatomischen, histologischen und molekularen Tumormerkmale klassifiziert.
2021 hat die WHO wichtige Änderungen an der Klassifizierung von Ependymomen vorgenommen und neue molekulare Merkmale genannt, die für die Prognose und patientenspezifische Therapieansätze von Bedeutung sind.
Molekulare Daten ergänzen die traditionelle histologische Klassifikation von Ependymomen erheblich und ermöglichen eine personalisierte Behandlung sowie eine Verbesserung der Prognose.
Anatomische Klassifikation mit molekularen Eigenschaften (WHO 2021)
Nach der WHO-Klassifikation der ZNS-Tumoren (2021) werden insgesamt zehn Ependymomsubtypen definiert:
Kommt seltener vor, eine bessere Prognose, häufig bei Säuglingen
Supratentorielles Ependymom o. n. A. (ST-EPN-NOS/NEC)
2 oder 3
Heterogene Mutationen, nicht-klassifiziert (NEC) und ohne molekulare Präzisierung (NOS)
Heterogene Gruppe, für Prognose und Therapie sind zusätzliche Untersuchungen erforderlich
Ependymom der hinteren Schädelgrube, Gruppe A (PF-EPN-A)
–
H3K27me3-Deletion; häufig 1q+
Aggressiv, schlechte Prognose, kommt vorwiegend bei Säuglingen und Kindern vor
Ependymom der hinteren Schädelgrube, Gruppe B (PF-EPN-B)
–
CIN, Aufrechterhaltung von H3K27me3
Gute Prognose, kommt bei Erwachsenen und Jugendlichen vor
Ependymom der hinteren Schädelgrube o. n. A. (PF-EPN-NOS/NEC)
2 oder 3
Morphologisch Ependymom der hinteren Schädelgrube, nicht-klassifiziert (NEC) und ohne molekulare Präzisierung (NOS)
Diagnose anhand der Lokalisation und der klassischen histologischen Merkmale. Die Prognose ist patientenspezifisch und hängt vom Resektionsgrad und der klinischen Dynamik ab
Spinales Ependymom
2 oder 3
Ohne eindeutige molekulare Marker, häufig 22q-Deletionen (in der NF2-Lokalisation)
Häufiger im kaudalen Abschnitt des Rückenmarks, lokale Rezidive, Genesung nach der Resektion und Strahlentherapie möglich
Subependymom (SubEPN)
1
I. d. R. ohne aggressive Mutationen, gutartig, ggf. mit TERT-Mutation
Beliebiger Abschnitt des Ventrikelsystems und des Rückenmarks, langsames Wachstum, häufig Zufallsbefund, gute Prognose
Bestimmung des Malignitätsgrades WHO (WHO Grad)
Nach der Bösartigkeit werden Ependymome in der Regel dem WHO Grad II oder III zugeordnet. Je höher der WHO-Grad ist, desto höher ist die Bösartigkeit des Tumors und desto schlechter ist die Prognose für den Patienten.
Nach der 2021 aktualisierten WHO-Klassifikation der Tumoren des zentralen Nervensystems werden neue Molekulartypen von Ependymomen wie YAP1, Fusion-positiv (ST-EPN-YAP1), ZFTA, Fusion-positiv(ST-EPN-ZFTA), PF-A und PF-B keinem Grad zugeordnet. Der Grad (Malignitätsgrad – Grad 1, 2 oder 3) sollte weiterhin nur für klassische (einschl. morphologisch definierter, aber ohne molekulare Präzision — „NEC/NOS“) Ependymome angegeben werden.
Definitionen für NOS und NEC:
NOS (Not Otherwise Specified) — „nicht anders spezifiziert, o. n. A.“: Dieser Begriff wird verwendet, wenn für den Tumor nicht alle erforderlichen zusätzlichen molekularen/genetischen oder anderen Tests durchgeführt wurden (oder durchgeführt werden können), sodass die Diagnose nur auf Grundlage der grundlegenden (morphologischen) Kriterien gestellt wird. Am Beispiel des Ependymoms: Wenn der Hirntumor der klassischen Histologie des Ependymoms entspricht, aber die molekulare Untergruppe (zum Beispiel ZFTA-Fusion, YAP1-Fusion, PF-A, PF-B) nicht bestimmt wurde oder die Analyse nicht geeignet ist, um eine genaue Diagnose zu stellen, schreibt man „supratentorielles Ependymom, NOS“ oder „Ependymom der hinteren Schädelgrube, NOS“.
NEC (Not Elsewhere Classified) — „nicht anderswo klassifiziert“: Dieser Begriff wird verwendet, wenn der Tumor vollständig beschrieben ist, alle modernen Tests durchgeführt wurden, aber er lässt sich keiner der bekannten und standardisierten Subtypen oder Gruppen zuordnen. Im Fall des Ependymoms: Wenn der Tumor den Kriterien für ein Ependymom entspricht, eine umfassende morphologische und molekulare Untersuchung durchgeführt wurde, jedoch nicht in einen der beschriebenen molekularen Subtypen passt (zum Beispiel nicht zu ZFTA, YAP1, PF-A oder PF-B), wird die Diagnose „Ependymom, NEC“ lauten.
Das Ependymom streut innerhalb des zentralen Nervensystems und verbreitet veränderte Zellen über die zirkulierende Liquorflüssigkeit.
Diagnostik der Ependymome
Magnetresonanztomographie (MRT)
Die Grundlage der Diagnose ist Gehirn- und Rückenmark-MRT mit Kontrastmittel.
Supra- und infratentorielle Ependymome enthalten normalerweise Verkalkungen und zystische Komponenten. Sie weisen Blutungen und ein unregelmäßig verstärktes MRT-Signal auf.
In der Bildgebung sind Ependymome in der Regel hypointens in T1 und hyperintens in T2, oft mit ausgeprägter Kontrastverstärkung.
Spinale Ependymome sind seltener verkalkt und können ein hypointenses T2-Signal ergeben, bedingt durch Hämosiderinablagerungen – das sogenannte „Cap Sign“ (Kappenzeichen).
Myxopapilläre Ependymome sind typischerweise isointens in T1 und hyperintens in T2, verglichen zu den spinalen Tumoren.
Liquorzytologie
Die Liquorzytologie (zytopathologische Untersuchung des Liquor cerebrospinalis) ist wichtig für das Grading und erfolgt in der Regel postoperativ (u. a. mit dem Ziel, das Ausmaß der nachfolgenden Strahlentherapie zu präzisieren).
Histologische Untersuchung
Bei der Tumorresektion ist der histologische Nachweis obligat. Das intraoperativ entnommene Tumormuster wird mikroskopisch untersucht, um seinen Typ und Grad der Malignität zu bestimmen.
Die chirurgische Entfernung ist die wirkungsvollste Therapie für Ependymome.
Das Ausmaß der Resektion ist von entscheidender Bedeutung für die Prognose. Der Eingriff erfordert eine hohe Qualifikation aufgrund der Nähe zu lebenswichtigen Strukturen.
Manchmal kann bei Hydrozephalus ein Shunt zur Ableitung des überschüssigen Liquors aus den Ventrikeln gelegt werden.
Die Lokalisation erschwert eine totale Entfernung von Ependymomen (es besteht ein hohes Komplikationsrisiko). In diesen Fällen steigt die Notwendigkeit einer postoperativen Strahlentherapie.
Strahlentherapie
Die postoperative Strahlentherapie hat einen relevanten Einfluss auf das progressionsfreie Überleben.
In den meisten Fällen ist sie nach subtotaler Entfernung des Tumors angezeigt.
Bei Streuung wird kraniospinale Bestrahlung empfohlen.
Spezielle Formen der Strahlentherapie (IMRT, Protonentherapie) minimieren den Schaden an gesundem Gewebe.
Chemotherapie
Chemotherapie ist nur begrenzt wirksam und wird hauptsächlich bei Kindern unter 1–1,5 Jahren verwendet, um die nebenwirkungsreiche Strahlentherapie möglicherweise hinauszuschieben, oder bei Rezidiven, wenn eine Operation oder Bestrahlung nicht möglich ist. Es kommen Cisplatin, Carboplatin, Cyclophosphamid, Etoposid und Methotrexat zum Einsatz.
Bei Kindern über 1 Jahr und Erwachsenen ist die Bedeutung der Chemotherapie aufgrund der geringen Empfindlichkeit von Ependymomen gegenüber den meisten Zytostatika eingeschränkt. In seltenen Fällen kann sie bei Rezidiven oder wenn chirurgische und strahlentherapeutische Maßnahmen nicht möglich sind, empfohlen werden.
Zielgerichtete Tumortherapie und Immuntherapie
Es gibt experimentelle Daten zur zielgerichteten Therapie anhand des molekularen Profils (z. B. EGFR- und VEGF-Inhibitoren), jedoch ist ihre klinische Anwendung begrenzt und erfordert weitere Forschungen.
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Prognose und Überwachung
Die Überlebensrate hängt vom Alter des Patienten, dem Resektionsgrad, dem molekularen Subtyp und der Dissemination (Streuung) des Tumors ab.
Bei kompletter Resektion und angemessener Therapie beträgt die 5-Jahres-Überlebensrate bei Kindern über 70%.
Bei erwachsenen Patienten hängt die Prognose ebenfalls vom Tumortyp ab und ist insgesamt günstig bei totaler Entfernung und anschließender Strahlentherapie.
Eine langfristige Überwachung mit regelmäßigen MRT-Untersuchungen von mindestens 5 Jahren ist angesagt, um mögliche Spätrezidive zu erkennen.
FAQ
1. Was ist ein Ependymom und wo kann es lokalisiert sein?
Bei Ependymomen handelt es sich um Gliome des zentralen Nervensystems, die aus den die Hirnhöhlen und den Rückenmarkskanal auskleidenden Ependymzellen entstehen. Bei Kindern sind bis zu 90% der Ependymome im Gehirn (insbesondere in der hinteren Schädelgrube) lokalisiert, wobei bei Erwachsenen 65% der Fälle spinale Ependymome ausmachen.
2. Welche Symptome sind für Ependymome typisch?
Das klinische Erscheinungsbild hängt direkt von der Lokalisation des Tumors ab. Bei Hirntumoren können Zeichen eines erhöhten Hirndrucks (Kopfschmerzen, Übelkeit), Krampfanfälle, Ataxie (Koordinationsstörungen) und fokale neurologische Defizite vorliegen. Für spinale Ependymome, einschließlich der Tumoren der Cauda equina, sind Rückenschmerzen, Muskelschwäche in den Beinen und Funktionsstörungen der Beckenorgane (Miktions- und Defäkationsstörungen) charakteristisch.
3. Sind Ependymome maligne oder benigne Tumoren?
Ependymome stellen eine breite Gruppe von Tumoren dar. Es gibt langsam wachsende, gutartige Formen (z. B. Subependymome, WHO Grad I), aber die meisten Ependymome gehören zu Grad II–III und werden als bösartig eingestuft. Die moderne WHO-Klassifikation basiert auf molekularen Eigenschaften, die den Malignitätsgrad und Prognose präzisieren.
4. Wie werden Ependymome behandelt?
Die wirkungsvollste Therapie für Ependymome ist ihre totale chirurgische Resektion. Da eine radikale Entfernung des Tumors nicht immer möglich ist, ist der zweite entscheidende Schritt die postoperative Strahlentherapie, die die Kontrolle über die Erkrankung erheblich verbessert. Die Rolle der Chemotherapie ist begrenzt: sie wird hauptsächlich bei Kleinkindern eingesetzt, um die Strahlentherapie möglicherweise hinauszuschieben, sowie bei Rezidiven, wenn andere Behandlungsoptionen keine Wirkung zeigen.
5. Wie ist die Prognose und die Überlebensrate bei Ependymomen?
Die Prognose und die Überlebensrate sind sehr variabel und hängen vom molekularen Subtyp des Tumors, dem Alter des Patienten und vor allem vom Resektionsgrad ab. Bei totaler Resektion und angemessener Therapie beträgt die 5-Jahres-Überlebensrate bei Kindern über 70%. Die Prognose bei Erwachsenen ist ebenfalls insgesamt günstig, vorausgesetzt, die Behandlung ist angemessen. Jedoch haben aggressive molekulare Subtypen wie PF-EPN-A bei Kindern oder MYCN-amplifizierte spinale Ependymome eine schlechtere Prognose.
Quellenverweise
1.
VOKA-Katalog. [Elektronische Quelle].
https://catalog.voka.io/
2.
Upadhyaya SA, Tinkle C. Intracranial ependymoma and other ependymal tumors. UpToDate. 2023
3.
Kresbach C, Neyazi S, Schüller U. Updates in the classification of ependymal neoplasms: The 2021 WHO Classification and beyond. Brain Pathol. 2022 Jul;32(4):e13068. doi: 10.1111/bpa.13068. Epub 2022 Mar 21. PMID: 35307892; PMCID: PMC9245931.
4.
Mu W, Dahmoush H. Classification and neuroimaging of ependymal tumors. Front Pediatr. 2023 May 23;11:1181211. doi: 10.3389/fped.2023.1181211. PMID: 37287627; PMCID: PMC10242666.
5.
Mack SC, Witt H, Pajtler KW et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. 2018;553(7686):101-5.
6.
Parker M et al. C11orf95-RELA fusions drive oncogenic NF-kB signalling in ependymoma. Nature. 2014;506(7489):451-5.
7.
Yamaguchi J et al. Latest classification of ependymoma in the molecular era and advances in its treatment: a review. Jpn J Clin Oncol. 2023;53(8):653-663.
8.
Merchant TE et al. Conformal radiotherapy after surgery for paediatric ependymoma: overall survival and toxicity. Lancet Oncol. 2009;10(3):258-66.
9.
National Comprehensive Cancer Network (NCCN) CNS Cancers Guidelines, 2024.
10.
Tsang DS et al. Reirradiation for recurrent pediatric intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2018;100(2):507-514.
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io