[{"title":"Galaktozele: \u00c4tiologie, Klinik, Diagnose und Behandlung","link":"https:\/\/wiki.voka.io\/de\/krankheit\/onkologie\/galaktozele\/","category_name":"Onkologie","is_video":"yes","image":"https:\/\/storage.googleapis.com\/dev_wiki_voka_io_303011\/articles\/en\/oncology\/galactocele\/3d-model-of-galactocele.webp"},{"title":"Fibroadenom der Brust: \u00c4tiologie, Klinik, Diagnose und Behandlung","link":"https:\/\/wiki.voka.io\/de\/krankheit\/onkologie\/fibroadenom\/","category_name":"Onkologie","is_video":"yes","image":"https:\/\/storage.googleapis.com\/dev_wiki_voka_io_303011\/articles\/en\/oncology\/fibroadenoma\/3d-model-of-fibroadenoma.webp"},{"title":"Brustzyste: \u00c4tiologie, Klassifikation, Klinik, Diagnose und Behandlung","link":"https:\/\/wiki.voka.io\/de\/krankheit\/onkologie\/brustzyste\/","category_name":"Onkologie","is_video":"yes","image":"https:\/\/storage.googleapis.com\/dev_wiki_voka_io_303011\/articles\/en\/oncology\/breast-cyst\/3d-model-of-breast-cyst.webp"}]
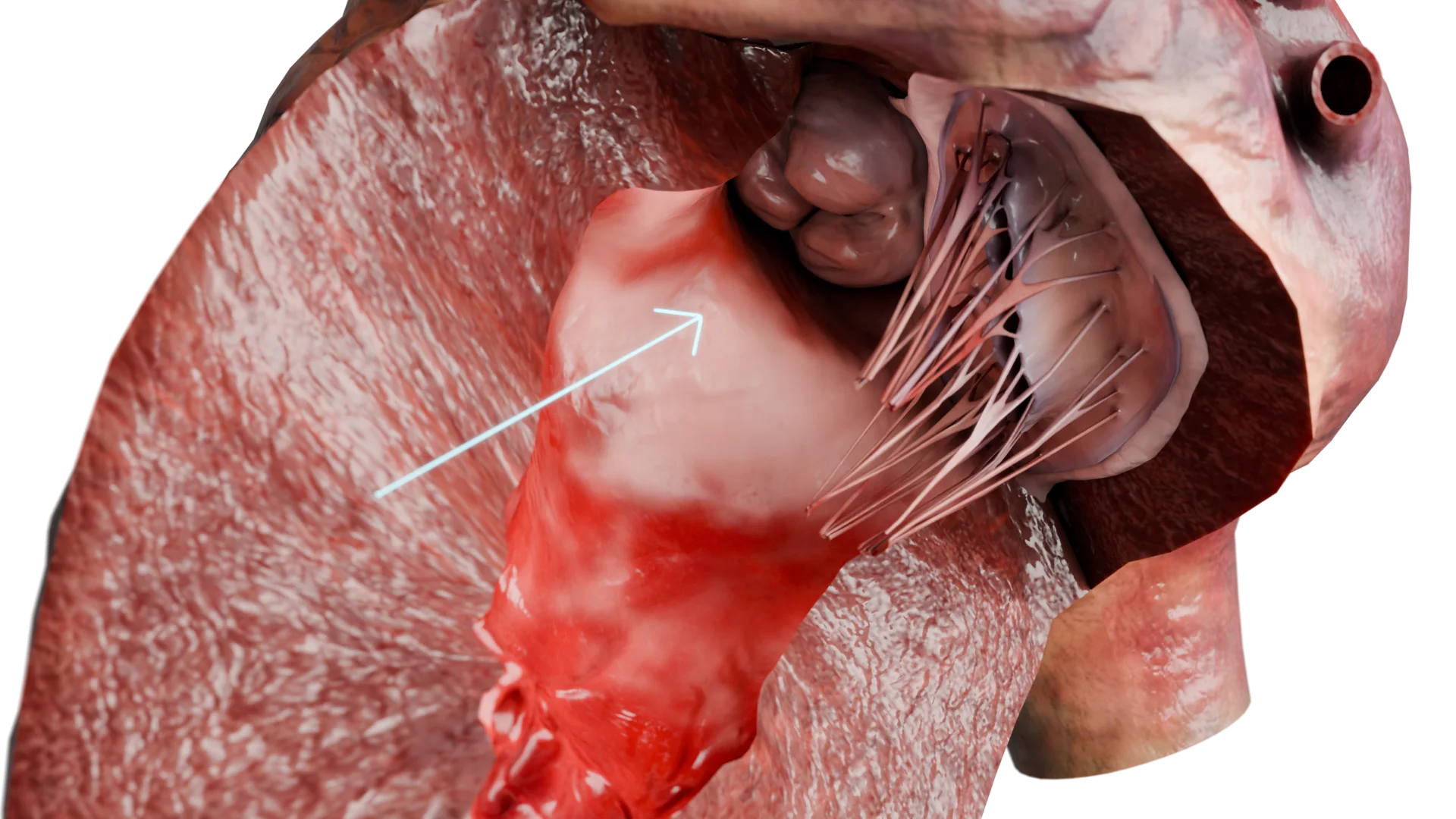
Die hypertrophe Kardiomyopathie (HCMP) ist eine primäre Herzmuskelerkrankung, die durch eine ungeklärte, meist asymmetrische Hypertrophie der linken Kammerwand ohne Vergrößerung der Herzkammer gekennzeichnet ist. Die Krankheit beruht auf Mutationen von Genen, die für sarkomerische Proteine kodieren, was zu morphologischen, elektrischen und hämodynamischen Anomalien führt.Linksventrikuläre Wandverdickung bei HCMP – 3D-Modell
Die HCMD ist eine der häufigsten vererbten Formen der Kardiomyopathie und tritt bei etwa 1 von 500 Erwachsenen auf. Männer sind häufiger betroffen als Frauen (ungefähres Verhältnis 3:2), aber Frauen werden in der Regel in einem späteren Alter diagnostiziert und haben eine schwerere Erkrankung.
Ätiologie
Die Krankheit beruht auf einer gestörten Synthese und Funktion sarkomerer kontraktiler Proteine, aber in einigen Fällen wurden sekundäre Formen im Zusammenhang mit systemischen und metabolischen Störungen festgestellt.
Bis zu 60-70 % der Fälle von hypertropher Kardiomyopathie sind monomutant genetisch bedingt.
Gene, die mit der Entwicklung von HCMD in Verbindung gebracht werden
Gen
Eiweiß
Häufigkeit von Mutationen
MYH7
Myosin β-schwere Kette
MYH7 und MYBPC3 machen etwa 70 % der Mutationsfälle aus
MYBPC3
Myosin-bindendes Protein C
MYH7 und MYBPC3 machen etwa 70 % der Mutationsfälle aus
TNNT2
Troponin T
Etwa 5 Prozent
TNNI3
Troponin I
<5%
TPM1
Tropomyosin
<5%
Die Krankheit wird autosomal-dominant vererbt, d. h. ein einziges verändertes Gen von einem der Elternteile reicht aus. In diesem Fall ist die Wahrscheinlichkeit, dass die Krankheit auftritt, hoch, aber die Schwere und die Form der Manifestationen können selbst innerhalb derselben Familie stark variieren.
In etwa 30 % der Fälle tritt die Mutation zum ersten Mal auf, ohne dass eine Familienanamnese vorliegt.
Einige Erkrankungen können das klinische Bild der hypertrophen Kardiomyopathie imitieren, haben aber einen anderen pathogenetischen Ursprung. Es ist äußerst wichtig, diese von der primären (sarkomeren) Form zu unterscheiden, da die Behandlung und die Prognose unterschiedlich sind.
Varianten mit sekundärer Hypertrophie (HCMP-Phänokopien)
Autosomal rezessive Krankheit, die durch Mutationen im FXN-Gen verursacht wird (mitochondriale Krankheit)
Fortschreitende Neurodegeneration (Ataxie, Muskelschwäche und -schwund, Sprachstörungen usw.).
Glykogenosen (z. B. Morbus Pompe)
Anhäufung von Glykogen in den Lysosomen der Zellen, insbesondere im Muskel und im Herzen
Häufig mit Beteiligung der Skelettmuskulatur
Systemische arterielle Hypertonie
Reaktive Myokardhypertrophie
Typischerweise symmetrisch, mit Bluthochdruck in der Vorgeschichte
Selbst bei Vorliegen einer Mutation im Sarkomerprotein-Gen hängen der klinische Schweregrad und der Verlauf von HCMP von weiteren Faktoren ab:
Epigenetische Regulatoren;
Assoziierte arterielle Hypertonie;
Intensive körperliche Betätigung (vor allem im Jugendalter);
Geschlecht und Hormonstatus (Frauen haben eher obstruktive Formen, die sich erst später manifestieren);
Ein plötzlicher Herztod in der Familie.
Pathogenese
Störung der sarkomeren Funktion
Mutationen in Genen (siehe Ätiologie) führen dazu:
Überempfindlichkeit gegen Kalzium;
Verminderte kontraktile Effizienz;
Erhöhter Energiebedarf.
Folge: Es entwickelt sich eine kompensatorische Myokardhypertrophie, vor allem des Interventrikelseptums, insbesondere im linken ventrikulären Ausflusstrakt (LVOP).
Hypertrophie und gestörte Entspannung (diastolische Dysfunktion)
Die Wandverdickung führt zu:
Verminderte linksventrikuläre Dehnbarkeit;
Störung der Füllung in der Diastole;
Erhöhter diastolischer Druck.
Folge: Entwicklung eines Staus im kleinen Kreislauf, der sich durch Dyspnoe und andere Symptome einer Herzinsuffizienz mit erhaltener Auswurffraktion bemerkbar macht.
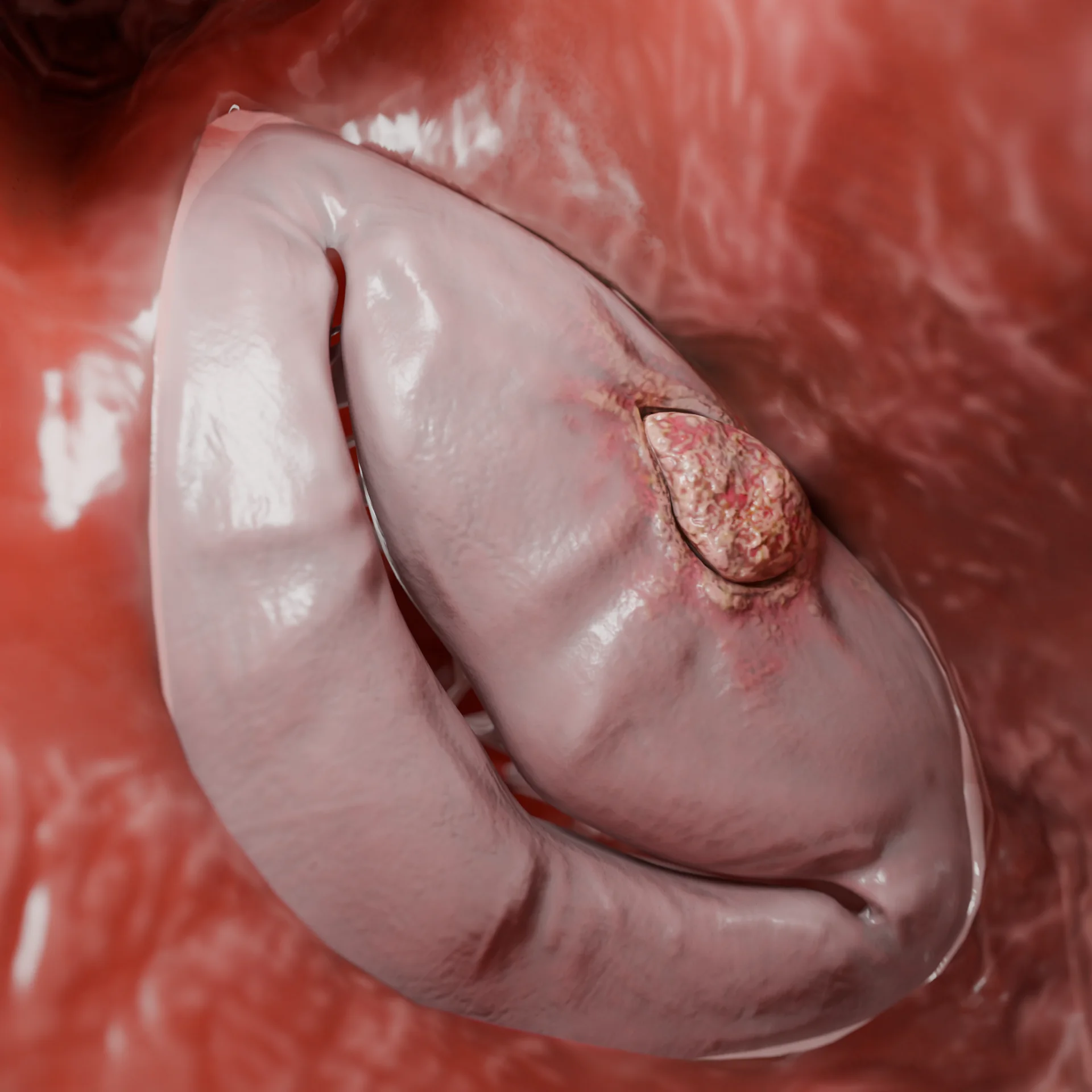
Starke Verengung des LV-Ausflussweges (obstruktive Form)
Bei 60-70 % der Patienten kommt es zu einer VTE-Obsistenz.
Aufgrund des Venturi-Effekts während der Systole kommt es zu einer Retraktion des vorderen Mitralklappenblättchens zum Septum (SAM-Phänomen), einer Vergrößerung des Druckgradienten und der Entwicklung einer Mitralinsuffizienz.
Die Folge: Die hämodynamische Überlastung nimmt zu, was die Symptome verschlimmert und das Risiko von Herzrhythmusstörungen erhöht.
VTE-Obstruktion durch Verdickung des Septums interventrikulare – 3D-Modell
Mikrovaskuläre Ischämie
Das hypertrophierte Myokard benötigt mehr Sauerstoff, aber:
Das Kapillarnetz hat keine Zeit, das Gewebewachstum zu kompensieren;
Es besteht ein Ungleichgewicht zwischen Sauerstoffbedarf und -zufuhr;
Häufig wird eine Fibrose der kleinkalibrigen Gefäße festgestellt.
Die Folge: In normalen Herzkranzgefäßen entwickelt sich eine Myokardischämie, die zu Brustschmerzen, Fibrose und einem erhöhten Risiko für Herzrhythmusstörungen führt.
Fibrose und elektrische Instabilität
Als Reaktion auf eine Myokardischämie und mechanische Überlastung bilden sich interstitielle und fokale Fibrosen, die zu einer Fibrose führen:
Störung der Impulsleitung;
Die Entwicklung von Herzrhythmusstörungen;
Erhöhung des Risikos eines plötzlichen Herztodes (SCD).
3D-Animation – hypertrophe Kardiomyopathie
Symptome
Dyspnoe bei Anstrengung, Müdigkeit;
Schmerzen in der Brust (Angina pectoris) ohne koronare Herzkrankheit;
Synkope oder Prä-Synkope (insbesondere bei Bewegung);
Herzrhythmusstörungen, Vorhofflimmern;
VSS – insbesondere bei jungen Patienten und Sportlern mit obstruktiver Form;
Herzinsuffizienz: kann mit erhaltener Auswurffraktion (HFpEF) oder mit einer Verringerung der Auswurffraktion im Spätstadium auftreten. Sie äußert sich durch Ödeme, Orthopnoe, Tachykardie und verminderte körperliche Belastbarkeit;
Asymptomatischer Verlauf: Bei 25-30 % der Patienten wird die Krankheit durch ein Screening der Familiengeschichte entdeckt. Dies schließt ein hohes Risiko für Komplikationen nicht aus.
Diagnose der hypertrophen Kardiomyopathie
Die Echokardiographie (transthorakale Echokardiographie) ist eine wichtige Methode der Primärdiagnose. Sie ermöglicht:
Beurteilung der Myokarddicke. Die Diagnose ist wahrscheinlich, wenn die Wanddicke ≥15 mm bei Erwachsenen oder ≥13 mm bei Verwandten in der ersten Linie mit bestätigter cGMP beträgt;
Definieren Sie die Verteilung der Hypertrophie: asymmetrisch, konzentrisch, apikal;
Nachweis einer LV-Ausflussbahnobstruktion (Gradient ≥30 mmHg, klinisch signifikant ≥50 mmHg);
Erkennung des SAM-Phänomens (systolische Retraktion des Mitralblättchens) und der Mitralinsuffizienz;
Beurteilung der LV-Funktion und des Vorliegens einer diastolischen Dysfunktion;
Messen Sie die Größe der Vorhöfe (insbesondere des linken Vorhofs – Risiko von FP).
Eine kardiale MRT wird empfohlen, wenn:
Mit EchoCG lässt sich die Wanddicke nicht genau beurteilen;
Es besteht der Verdacht auf eine apikale oder atypische Form von HCMP;
Eine Bewertung der Myokardfibrose ist erforderlich.
Enthüllt:
Verteilung und Grad der Hypertrophie;
Fibrosebereiche – verbunden mit einem erhöhten Risiko für Herzrhythmusstörungen und VSS;
Differenzierung mit Phänokopien (z. B. Amyloidose).
EKG (Elektrokardiographie) – unspezifisch, aber pathologische Veränderungen werden bei mehr als 90 % der Patienten festgestellt. Anzeichen einer linksventrikulären Hypertrophie:
Atypische Q-Zähne (in den Ableitungen V4-V6, I, aVL) – können einen Myokardinfarkt vortäuschen;
ST-Segmentveränderungen und Inversion der T-Wellenform;
Bewertung des Schweregrads von Rhythmusstörungen und Indikationen für ICD.
Ein Belastungstest (Laufband oder Fahrradergometrie). Gehalten für:
Bewertung der Belastungstoleranz;
Erkennung von induzierbaren VTE-Verschlüssen;
Bestimmung der Steigung gegen die Last;
Erkennung von Koronarsymptomen ohne Koronarstenose.
Genetische Tests. Empfohlen:
Patienten mit bestätigter cGMP (insbesondere jüngere Patienten oder solche mit HCC in der Familiengeschichte);
Zur Untersuchung von Verwandten in erster Linie (Kinder, Geschwister, Eltern);
Bei Verdacht auf Phänokopien (Morbus Fabry, mitochondriale Erkrankungen usw.).
Identifizierte Gene: MYH7, MYBPC3, TNNT2, TNNI3, TPM1 sind die häufigsten Gene.
Labor-Marker:
NT-proBNP / BNP – erhöht bei Drucküberlastung, diastolischer Dysfunktion;
Troponin T/I – kann bei mikrovaskulärer Ischämie mäßig erhöht sein.
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Behandlung der hypertrophen Kardiomyopathie
Änderung des Lebensstils:
Ausschluss von schwerer körperlicher Betätigung und Sport;
Kontrolle von Blutdruck und Körpergewicht;
Screening von Verwandten in erster Linie.
Medikamentöse Therapie:
β-Blocker (vorzugsweise: Bisoprolol, Metoprolol);
Verapamil – im Falle von Gegenanzeigen für β-Blocker;
Disopyramid – als Zusatztherapie bei der obstruktiven Form;
Bei Mavacamten handelt es sich um ein neues Medikament mit nachgewiesener Wirkung zur Gradientensenkung und Symptomverbesserung (gemäß ESC 2023 und AHA 2020).
Chirurgische Behandlung
Indikationen:
Linksventrikulärer Ausflusstraktgradient (LVOG) ≥50 mmHg in Ruhe oder bei Provokation;
Schwere Symptome (NYHA III-IV), die nicht auf eine Behandlung mit β-Blockern, Verapamil oder Disopyramid ansprechen;
Ausgeprägte Mitralinsuffizienz in Verbindung mit dem SAM-Phänomen.
Erweiterte Septum-Myektomie:
Der Goldstandard der Chirurgie bei obstruktiver cGMP;
Sie wird durch eine Mininotomie oder eine vollständige Sternotomie durchgeführt, in einigen Fällen auch durch eine anteriore rechte Minithorakotomie;
Ein Teil der hypertrophierten Kammerscheidewand wird entfernt, wodurch der Druckgradient beseitigt wird;
Falls erforderlich, wird eine Mitralklappenplastik oder eine Resektion der Sekundärchordae durchgeführt;
Mögliche Komplikationen: Verstopfungen, Notwendigkeit eines ICD, Wiederauftreten des Gradienten.
Die Alkoholseptumablation (endovaskulärer Eingriff) wird häufiger bei Patienten eingesetzt, bei denen eine offene Operation kontraindiziert oder mit zu hohen Risiken verbunden ist.
Ethanolinjektion in die Perforansarterie → lokaler Infarkt → Verringerung der Septumdicke;
Risiko einer vollständigen AV-Blockade (bis zu 10 %) – Bereitschaft zur ECS-Implantation ist erforderlich;
Weniger Vorhersehbarkeit der Ergebnisse;
Die Möglichkeit einer unvollständigen Entfernung der Obstruktion.
Implantation eines Kardioverter-Defibrillators (ICD)
Indikationen:
Erfahrung mit VSS oder anhaltender VT;
LV-Wanddicke >30 mm;
VSS-Familiengeschichte;
Synkope unklarer Ätiologie;
LWF <50% im progressiven Verlauf.
Herztransplantation bei Patienten mit terminaler, refraktärer KHK trotz Behandlung.
FAQ
1. Was ist hypertrophe Kardiomyopathie?
Die hypertrophe Kardiomyopathie (HCMP) ist eine Erkrankung, bei der der Herzmuskel (in der Regel die Scheidewand zwischen den Herzkammern) abnorm verdickt ist. Im Gegensatz zur Hypertrophie aufgrund von Bluthochdruck oder Herzklappenfehlern ist die Verdickung bei der HCMP auf genetische Mutationen zurückzuführen und nicht auf ein überlastetes Herz.
2. Wird cGMP vererbt? Ist es notwendig, Verwandte zu untersuchen?
Ja, in den meisten Fällen wird HCMP autosomal dominant vererbt. Es wird empfohlen, die Verwandten in erster Linie zu untersuchen.
3. Welche Symptome können auf HCMP hinweisen und wann sollte ich einen Arzt aufsuchen?
Zu den Symptomen gehören Kurzatmigkeit, Schmerzen in der Brust, Schwindel, Ohnmacht, insbesondere bei Anstrengung. Wenn Sie diese Beschwerden oder eine familiäre Vorgeschichte mit plötzlichem Tod haben, sollten Sie einen Kardiologen aufsuchen.
4. Ist HCMP gefährlich? Ist es möglich, lange damit zu leben?
Bei rechtzeitiger Diagnose und angemessener Behandlung können die meisten Patienten ein erfülltes Leben führen. In einigen Fällen erhöht die HCMR jedoch das Risiko von Herzrhythmusstörungen und plötzlichem Tod, insbesondere ohne Behandlung.
5. Wie unterscheidet sich obstruktives HCMP von nichtobstruktivem cGMP?
Bei der obstruktiven Form behindert die verdickte Scheidewand den Abfluss des Blutes aus der linken Herzkammer, was schwerere Symptome verursacht. Bei der nicht-obstruktiven Form ist der Abfluss nicht beeinträchtigt.
6. Kann ich Sport treiben, wenn ich eine HCMP habe?
Intensiver und wettkampforientierter Sport wird bei HCMP nicht empfohlen. Moderate körperliche Aktivität ist in Absprache mit dem behandelnden Arzt erlaubt.
7. Welche Untersuchungen sind erforderlich, um die Diagnose HCMP zu stellen?
In der Regel werden ein EKG, eine Echokardiographie (Herz-Ultraschall), ein Herz-MRT, eine tägliche EKG-Überwachung und Gentests (falls angezeigt) durchgeführt.
8. Wie wird HCMP behandelt: Ist ein chirurgischer Eingriff erforderlich oder reichen Tabletten aus?
Die medikamentöse Behandlung ist die Hauptstütze der Behandlung. In schweren Fällen kann eine Operation oder eine perkutane Septumablation erforderlich sein.
9. Was ist ein ICD und wann wird es in der HCMP eingesetzt?
Ein ICD ist ein implantierbarer Kardioverter-Defibrillator, ein Gerät, das den plötzlichen Tod durch Herzrhythmusstörungen verhindert. Er wird Patienten mit hohem Risiko auf Anraten eines Arztes eingesetzt.
10. Ist es möglich, HCMP vollständig zu heilen? Wie sieht die Prognose aus?
HCMP kann nicht vollständig geheilt werden, aber die Symptome können wirksam kontrolliert werden. Mit dem richtigen Ansatz ist die Prognose günstig, insbesondere wenn keine schweren Komplikationen auftreten.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.