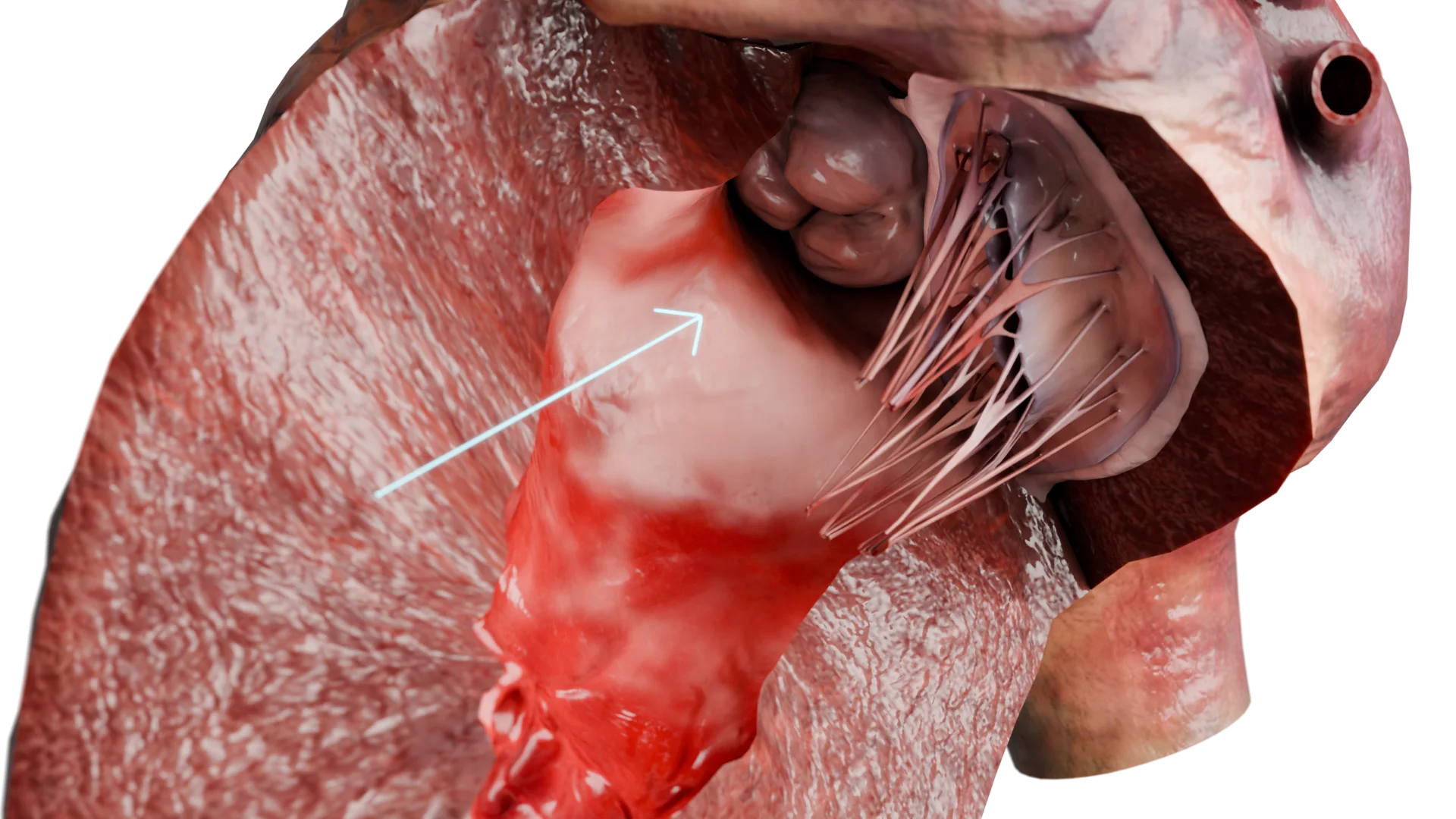
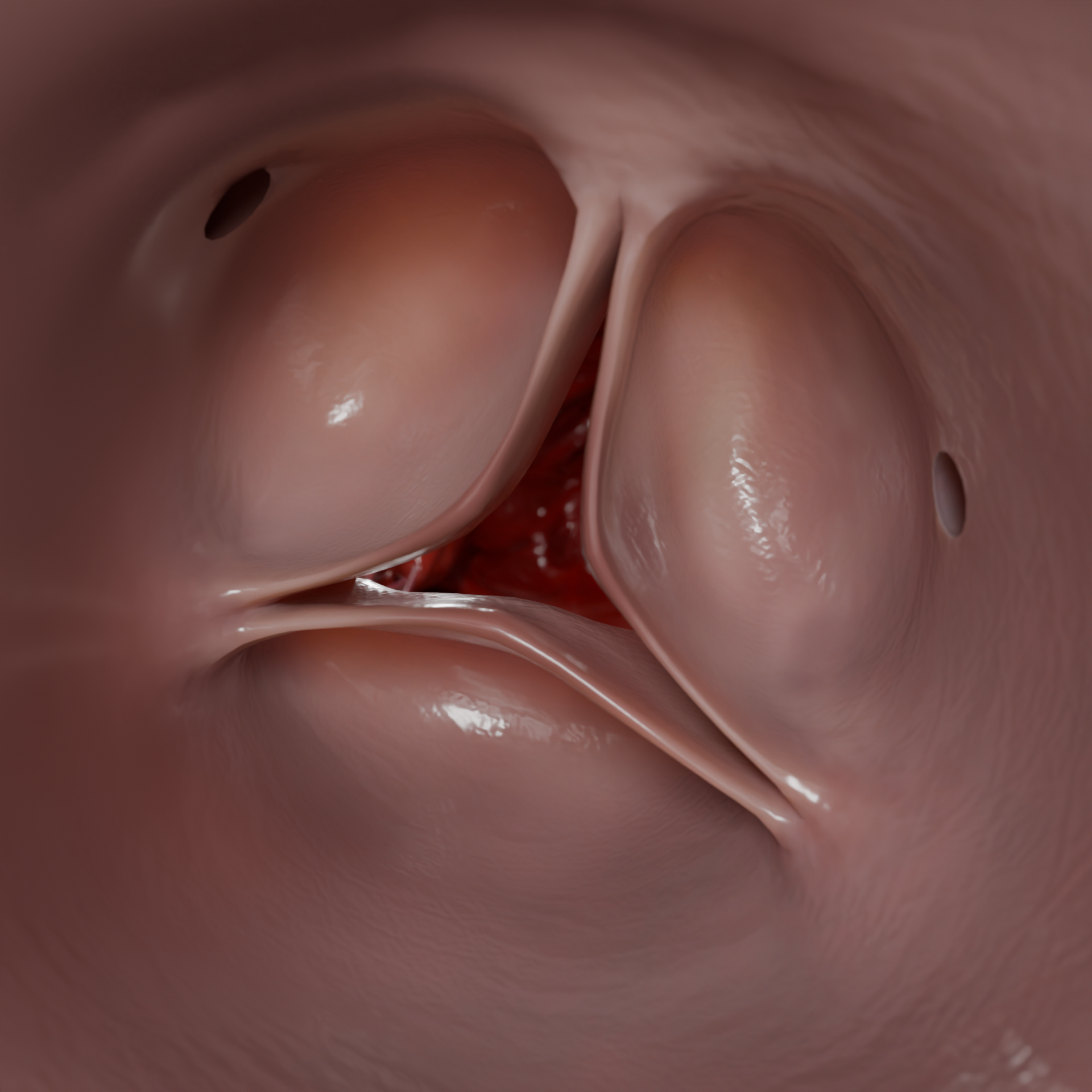
A cardiomiopatia hipertrófica (CMH) é uma doença primária do miocárdio caracterizada por hipertrofia inexplicada da parede ventricular esquerda, na maioria das vezes assimétrica, sem aumento da cavidade. A doença baseia-se em mutações dos genes que codificam as proteínas sarcoméricas, levando a anomalias morfológicas, eléctricas e hemodinâmicas.Espessamento da parede do ventrículo esquerdo no HCMP – Modelo 3D
A HCMD é uma das formas hereditárias mais comuns de cardiomiopatia e ocorre em cerca de 1 em cada 500 adultos. Os homens são mais frequentemente afectados do que as mulheres (proporção aproximada de 3:2), mas as mulheres são normalmente diagnosticadas numa idade mais avançada e têm uma doença mais grave.
A etiologia
A doença baseia-se na síntese e função deficientes das proteínas contrácteis sarcoméricas, mas em alguns casos foram identificadas formas secundárias associadas a perturbações sistémicas e metabólicas.
Até 60-70% dos casos de cardiomiopatia hipertrófica são de natureza genética monomutante.
Genes associados ao desenvolvimento da HCMD
Gene
Proteína
Frequência de mutação
MYH7
cadeia pesada da miosina β
MYH7 e MYBPC3 são responsáveis por cerca de 70% dos casos de mutação
MYBPC3
proteína C de ligação à miosina
MYH7 e MYBPC3 são responsáveis por cerca de 70% dos casos de mutação
TNNT2
Troponina T
Cerca de 5 por cento
TNNI3
Troponina I
<5%
TPM1
Tropomiosina
<5%
A doença é transmitida de forma autossómica dominante, ou seja, basta um único gene alterado de um dos progenitores. Neste caso, a probabilidade da doença é elevada, mas a gravidade e a forma das manifestações podem variar muito, mesmo dentro da mesma família.
Em cerca de 30% dos casos, a mutação ocorre pela primeira vez, sem história familiar.
Algumas doenças podem imitar o quadro clínico da cardiomiopatia hipertrófica, mas têm uma origem patogénica diferente. É extremamente importante distingui-las da forma primária (sarcomérica), uma vez que o tratamento e o prognóstico são diferentes.
Variantes com hipertrofia secundária (fenocópias do HCMP)
Doença
Mecanismo
Caraterísticas
Doença de Fabry
Doença hereditária de armazenamento lisossómico (deficiência da enzima α-galactosidase A)
Sinais de envolvimento sistémico (angioqueratomas, neuropatia, proteinúria)
Amiloidose do coração
Deposição de proteínas (AL, ATTR) no miocárdio
Espessamento da parede, redução da contratilidade, disfunção diastólica
Ataxia de Friedreich
Doença autossómica recessiva causada por mutações no gene FXN (doença mitocondrial)
Neurodegenerescência progressiva (ataxia, fraqueza e atrofia muscular, perturbações da fala, etc.).
Glicogenoses (por exemplo, doença de Pompe)
Acumulação de glicogénio nos lisossomas das células, especialmente no músculo e no coração
Frequentemente com envolvimento do músculo esquelético
Hipertensão arterial sistémica
Hipertrofia reactiva do miocárdio
Tipicamente simétrico, com história de hipertensão
Mesmo na presença de uma mutação no gene da proteína sarcomérica, a gravidade clínica e a evolução da HCMP dependem de factores adicionais:
Reguladores epigenéticos;
Hipertensão arterial associada;
Atividade física de alta intensidade (especialmente durante a adolescência);
Género e estado hormonal (as mulheres têm mais probabilidades de ter formas obstrutivas mas de manifestação mais tardia);
História familiar de morte súbita cardíaca.
Patogénese
Perturbação da função sarcomérica
As mutações nos genes (ver etiologia) provocam:
Hipersensibilidade ao cálcio;
Diminui a eficiência contrátil;
Aumento das necessidades energéticas.
Consequência: desenvolve hipertrofia miocárdica compensatória, predominantemente do septo interventricular, especialmente na via de saída do ventrículo esquerdo (VSVE).
Hipertrofia e relaxamento prejudicado (disfunção diastólica)
O espessamento da parede leva a:
Reduz a distensibilidade do ventrículo esquerdo;
Perturbação do seu enchimento na diástole;
Aumento da pressão diastólica.
Consequência: desenvolvimento de estase no pequeno círculo de circulação sanguínea, que se manifesta por dispneia e outros sintomas de insuficiência cardíaca com fração de ejeção preservada.
Estreitamento grave da via de saída do VE (forma obstrutiva)
Em 60-70% dos doentes, forma-se uma obstrução do TEV.
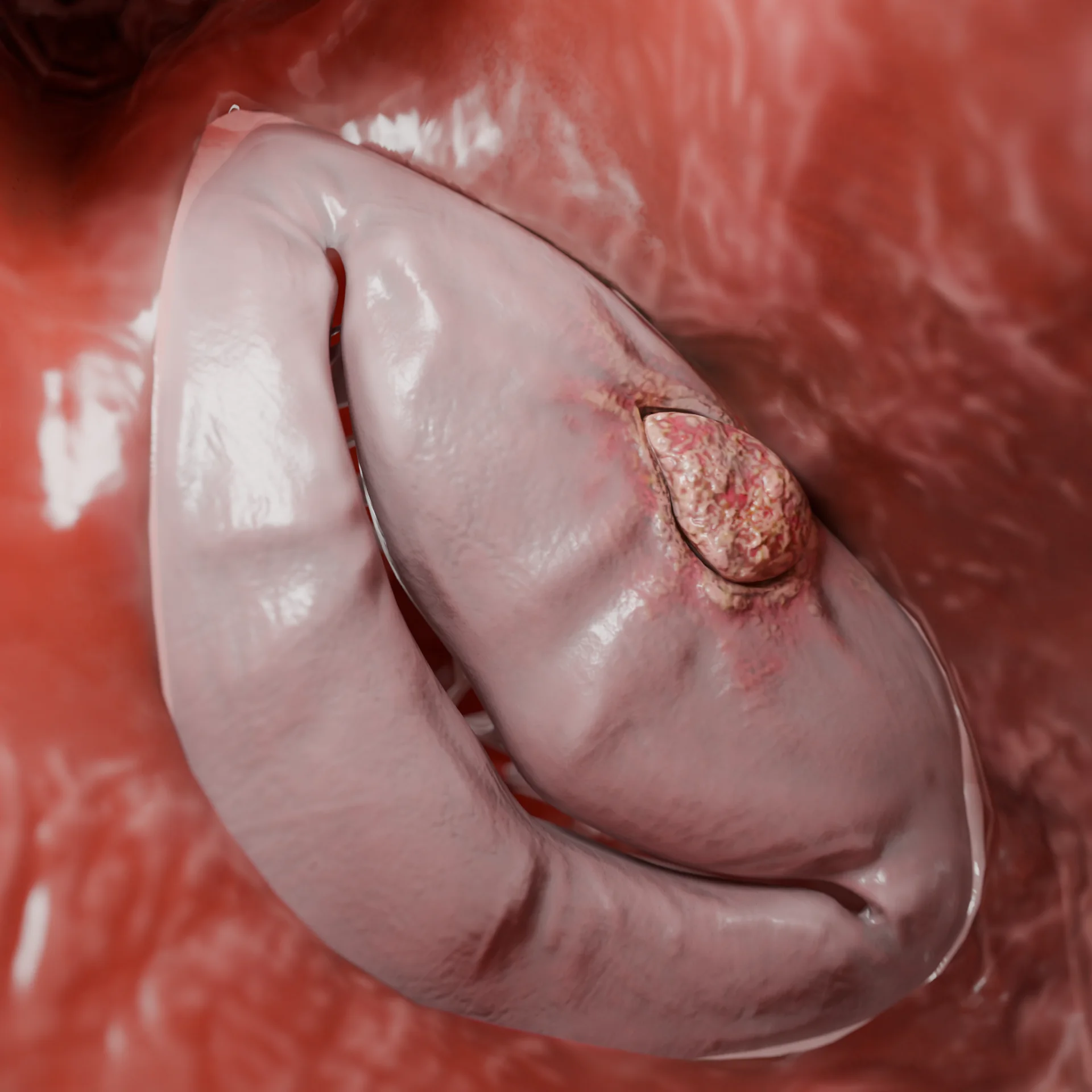
Devido ao efeito Venturi durante a sístole, ocorre: retração do folheto anterior da válvula mitral para o septo (fenómeno SAM), agravamento do gradiente de pressão, desenvolvimento de regurgitação mitral.
Consequência: aumenta a sobrecarga hemodinâmica, o que agrava os sintomas e aumenta o risco de arritmias.
Obstrução do TEV devido ao espessamento do septo interventricular – Modelo 3D
Isquémia microvascular
O miocárdio hipertrofiado necessita de mais oxigénio, mas:
A rede capilar não tem tempo para compensar o crescimento do tecido;
Existe um desequilíbrio entre a procura e o fornecimento de oxigénio;
É frequentemente detectada fibrose dos vasos de pequeno calibre.
Consequência: desenvolve-se isquémia do miocárdio em artérias coronárias normais, levando a dores no peito, fibrose e aumento do risco de arritmias.
Fibrose e instabilidade eléctrica
Em resposta à isquémia do miocárdio e à sobrecarga mecânica, forma-se fibrose intersticial e focal, resultando em
Distúrbio de condução de impulsos;
O desenvolvimento de arritmias ventriculares;
Aumenta o risco de morte súbita cardíaca (MSC).
Animação 3D – Cardiomiopatia hipertrófica
Manifestações clínicas
Dispneia ao esforço, fadiga;
Dor no peito (angina de peito) sem doença das artérias coronárias;
Síncope ou pré-síncope (especialmente com exercício);
Arritmias ventriculares, fibrilhação auricular;
VSS – especialmente em doentes jovens e atletas com forma obstrutiva;
Insuficiência cardíaca: pode ocorrer com fração de ejeção preservada (ICFEP) ou com a sua redução em fases tardias. Manifesta-se por edema, ortopneia, taquicardia, diminuição da tolerância ao exercício;
Evolução assintomática: em 25-30% dos doentes, a doença é detectada através do rastreio da história familiar. Não exclui um risco elevado de complicações.
Diagnóstico da cardiomiopatia hipertrófica
O ecocardiograma (ecocardiograma transtorácico) é um método fundamental de diagnóstico primário. Permite:
Avalia a espessura do miocárdio. O diagnóstico é provável quando a espessura da parede é ≥15 mm em adultos ou ≥13 mm em familiares de primeira linha com cGMP confirmado;
Define a distribuição da hipertrofia: assimétrica, concêntrica, apical;
Detecta a obstrução da via de saída do VE (gradiente ≥30 mmHg, clinicamente significativo ≥50 mmHg);
Detecta o fenómeno SAM (retração sistólica do folheto mitral) e a regurgitação mitral;
Avalia a função do VE e a presença de disfunção diastólica;
Mede o tamanho das aurículas (especialmente a aurícula esquerda – risco de FP).
A ressonância magnética cardíaca é recomendada se:
O EchoCG não avalia com exatidão a espessura da parede;
Suspeita-se de uma forma apical ou atípica de HCMP;
É necessária uma avaliação da fibrose miocárdica.
Revela:
Distribuição e grau de hipertrofia;
Áreas de fibrose – associadas a maior risco de arritmias e SAV;
Diferenciação com fenocópias (por exemplo, amiloidose).
ECG (eletrocardiografia) – não específico, mas são encontradas alterações patológicas em mais de 90% dos doentes. Sinais de hipertrofia do ventrículo esquerdo:
Dentes Q atípicos (nas derivações V4-V6, I, aVL) – podem simular infarto do miocárdio;
Alterações do segmento ST e inversão da onda T;
Perturbações do ritmo: fibrilhação auricular, extra-sístoles ventriculares, TV;
Bloqueio atrioventricular ou intraventricular.
Monitorização Holter. Indicações:
Suspeita de arritmias (VE, VT, FP);
Síncope ou pré-síncope;
Avaliação da gravidade dos distúrbios do ritmo e das indicações para CDI.
Uma prova de esforço (tapete rolante ou cicloergometria). Realizado para:
Avalia a tolerância ao exercício;
Deteção de obstrução induzível do TEV;
Determina o gradiente em função da carga;
Identificação de sintomas coronários sem estenose das artérias coronárias.
Testes genéticos. Recomenda-o:
Pacientes com cGMP confirmado (especialmente aqueles que são jovens ou têm uma história familiar de CHD);
Para rastrear familiares de primeira linha (filhos, irmãos, pais);
Em fenocópias suspeitas (doença de Fabry, doenças mitocondriais, etc.).
Genes identificados: MYH7, MYBPC3, TNNT2, TNNI3, TPM1 são os genes mais frequentes.
Marcadores laboratoriais:
NT-proBNP / BNP – elevado na sobrecarga de pressão, disfunção diastólica;
Troponina T/I – pode estar moderadamente elevada na isquémia microvascular.
Encontra mais conteúdos cientificamente exactos nas nossas redes sociais
Subscreve e não percas os recursos mais recentes
Tratamento da cardiomiopatia hipertrófica
Modificação do estilo de vida:
Exclusão de esforços físicos pesados e de desportos;
Controlo da tensão arterial e do peso corporal;
Rastreio de familiares de primeira linha.
Terapia medicamentosa:
β-bloqueadores (de preferência: bisoprolol, metoprolol);
Verapamil – em caso de contra-indicações para β-bloqueadores;
Disopiramida – como adjuvante na forma obstrutiva;
O Mavacamten é um novo medicamento com um efeito comprovado de redução do gradiente e melhoria dos sintomas (de acordo com a ESC 2023 e a AHA 2020).
Tratamento cirúrgico
Indicações:
Gradiente do trato de saída do ventrículo esquerdo (LVOG) ≥50 mmHg em repouso ou sob provocação;
Sintomas graves (NYHA III-IV) não passíveis de tratamento com β-bloqueadores, verapamil ou disopiramida;
Regurgitação mitral proeminente associada ao fenómeno SAM.
Miectomia septal alargada:
O padrão de ouro da cirurgia para o cGMP obstrutivo;
Realiza-se através de uma mininotomia ou esternotomia total, em alguns casos eventualmente através de uma minitoracotomia anterior direita;
Uma secção do septo interventricular hipertrofiado é removida, eliminando o gradiente de pressão;
Se necessário, é efectuada uma plastia da válvula mitral ou a ressecção das cordas secundárias;
Possíveis complicações: bloqueios, necessidade de CDI, recorrência do gradiente.
A ablação septal por álcool (intervenção endovascular) é mais frequentemente utilizada em doentes para os quais a cirurgia aberta está contra-indicada ou apresenta riscos demasiado elevados.
Injeção de etanol na artéria perfurante → infarto local → redução da espessura do septo;
Risco de bloqueio AV completo (até 10%) – é necessário estar preparado para o implante de ECS;
Menos previsibilidade dos resultados;
A possibilidade de remoção incompleta da obstrução.
Implantação de um cardioversor desfibrilhador (CDI)
Indicações:
Experimenta VSS ou VT sustentado;
Espessura da parede do VE >30 mm;
História da Família VSS;
Síncope de etiologia desconhecida;
LWF <50% no curso progressivo.
Transplante cardíaco em doentes com CH terminal e refratário apesar do tratamento.
FAQ
1. O que é a cardiomiopatia hipertrófica?
A cardiomiopatia hipertrófica (CMH) é uma doença em que o músculo cardíaco (normalmente o septo entre os ventrículos) fica anormalmente espessado. Ao contrário da hipertrofia causada pela hipertensão ou por defeitos nas válvulas, na CMHH o espessamento deve-se a mutações genéticas e não a uma sobrecarga do coração.
2. O cGMP é hereditário? É necessário examinar os teus familiares?
Sim, na maioria dos casos, o cGMP é herdado num padrão autossómico dominante. Recomenda-se que examines os familiares de primeira linha.
3. Que sintomas podem indicar cGMP e quando é que devo procurar assistência médica?
Os sintomas incluem falta de ar, dores no peito, tonturas, desmaios, especialmente quando fazes esforço. Se tiveres estas queixas ou uma história familiar de morte súbita, deves consultar um cardiologista.
4 As cGMP são perigosas? É possível viveres com ele durante muito tempo?
Com um diagnóstico atempado e um tratamento adequado, a maioria dos doentes vive uma vida plena. No entanto, em alguns casos, a HCMR aumenta o risco de arritmias e morte súbita, especialmente sem tratamento.
5. Em que é que o GMPc obstrutivo difere do GMPc não obstrutivo?
Na forma obstrutiva, o septo espessado interfere com o fluxo de sangue do ventrículo esquerdo, o que provoca sintomas mais graves. Na forma não obstrutiva, a saída de sangue não é afetada.
6. Posso fazer exercício quando tenho um cGMP?
Os desportos intensos e competitivos não são recomendados no cGMP. É permitida uma atividade física moderada, acordada com o médico assistente.
7. Que exames são necessários para fazer um diagnóstico de cGMP?
Normalmente, é realizado um ECG, ecocardiografia (ultrassom cardíaco), ressonância magnética cardíaca, monitorização diária do ECG e testes genéticos (quando indicado).
8. Como é que o cGMP é tratado: é necessária cirurgia ou bastam comprimidos?
A medicação é a base do tratamento. Em casos graves, pode ser necessário recorrer à cirurgia ou à ablação percutânea do septo.
9. O que é um CDI e quando é que é colocado no cGMP?
Um CDI é um desfibrilhador cardioversor implantável, um dispositivo que previne a morte súbita causada por arritmias. É inserido em pacientes de alto risco sob recomendação de um médico.
10. É possível curar completamente o cGMP? Qual é o teu prognóstico?
A CMP não pode ser completamente curada, mas os sintomas podem ser eficazmente controlados. Com a abordagem correta, o prognóstico é favorável, especialmente na ausência de complicações graves.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.