Cardiomiopatia hipertrófica: Etiologia, patogénese, sintomas, diagnóstico, métodos de tratamento
Kizyukevich O.Cirurgião cardiovascular, MD
14 min ler·Julho 22, 2025
Este artigo é apenas para fins informativos
O conteúdo deste sítio Web, incluindo texto, gráficos e outros materiais, é fornecido apenas para fins informativos. Não se destina a servir de conselho ou orientação. Relativamente ao teu estado de saúde ou tratamento específico, consulta o teu profissional de saúde.
Cardiomiopatia hipertrófica (CMH) é uma doença miocárdica primária caracterizada por hipertrofia inexplicada da parede do ventrículo esquerdo, geralmente assimétrica, sem dilatação da cavidade. A doença é causada por mutações em vários genes que codificam proteínas sarcoméricas, levando a anormalidades morfológicas, elétricas e hemodinâmicas.
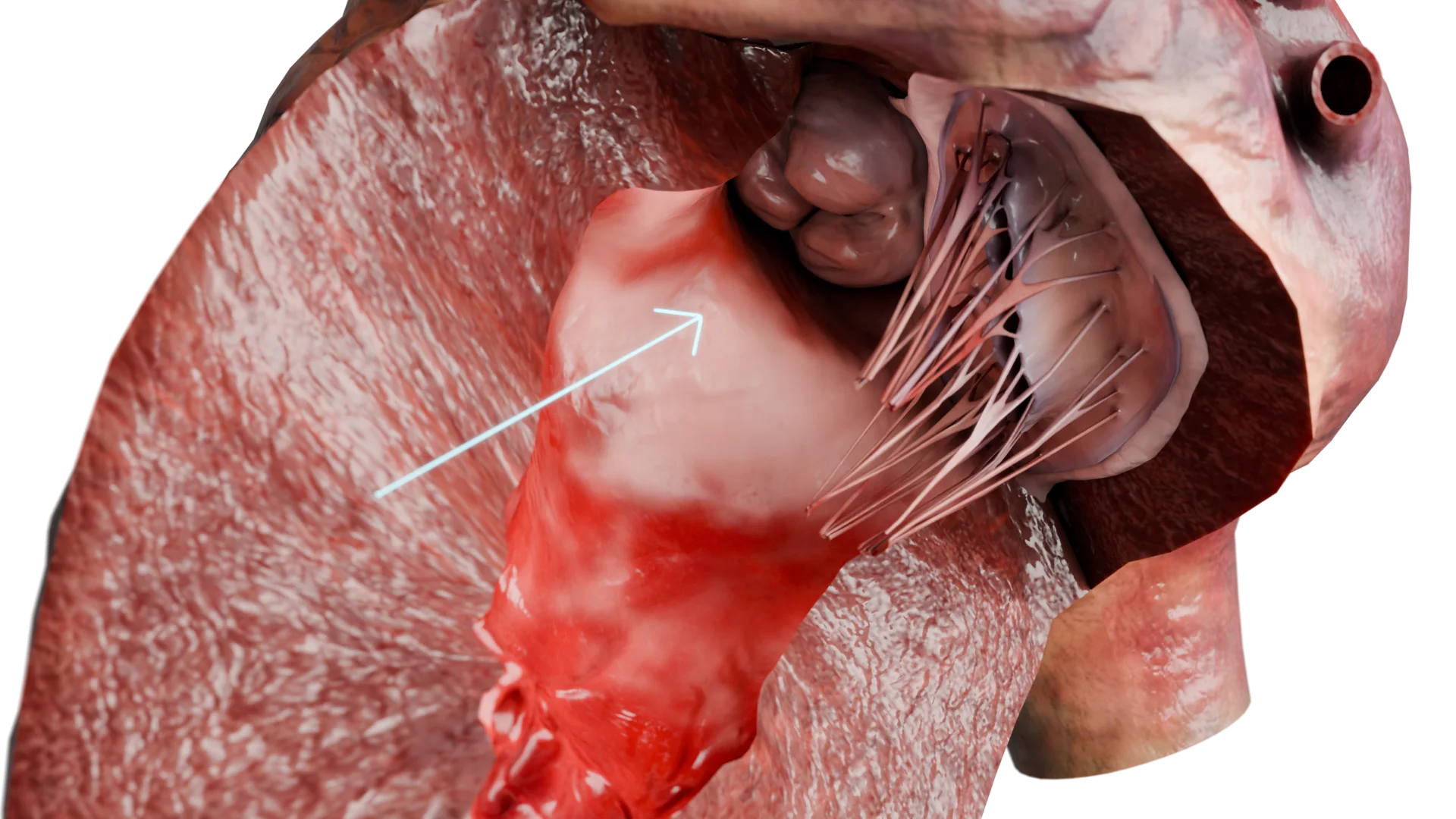
Espessamento da parede do ventrículo esquerdo na CMH — Modelo 3D
A CMH é uma das formas hereditárias mais comuns de cardiomiopatia, afetando aproximadamente 1 em cada 500 adultos. Os homens são afetados com mais frequência do que as mulheres (aproximadamente 3:2), mas nas mulheres, a doença geralmente é diagnosticada em idade mais avançada e é mais grave.
Etiologia
A doença é causada por uma alteração na síntese e na função das proteínas contráteis do sarcômero, mas, em alguns casos, são detectadas formas secundárias associadas a distúrbios sistêmicos e metabólicos.
Até 60-70% dos casos de cardiomiopatia hipertrófica têm natureza genética monomutante.
Genes associados ao desenvolvimento da CMH
Gene
Proteína
Frequência das mutações
MYH7
Cadeia pesada da beta miosina
O MYH7 e o MYBPC3 são responsáveis por cerca de 70% das mutações
MYBPC3
Proteína C ligante da miosina
O MYH7 e o MYBPC3 são responsáveis por cerca de 70% das mutações
TNNT2
Troponina Т
Aproximadamente 5%
TNNI3
Troponina I
<5%
TPM1
Tropomiosina
<5%
A doença é de padrão de herança autossômica dominante, o que significa que para desenvolvê-la, basta herdar uma cópia do gene alterado de um dos pais. Embora a probabilidade de desenvolver a doença seja alta, a gravidade e a forma das manifestações podem variar muito, mesmo dentro da mesma família.
Em aproximadamente 30% dos casos, a mutação se manifesta pela primeira vez, sem histórico familiar.
Algumas doenças podem mimetizar a apresentação clínica da cardiomiopatia hipertrófica, tendo, porém, uma origem patogenética diferente. É extremamente importante distingui-las da forma primária (sarcomérica), uma vez que o tratamento e o prognóstico são diferentes.
Variantes com hipertrofia secundária (fenótipos da miocardiopatia hipertrófica)
Doença
Mecanismo
Características
Doença de Fabry
Doença hereditária de depósito lisossômico (deficiência da enzima α-galactosidase A)
Sinais de envolvimento sistêmico (angioqueratoma, neuropatia, proteinúria)
Amiloidose cardíaca
Deposição de proteínas (AL, ATTR) no miocárdio
Espessamento da parede, diminuição da contratilidade, disfunção diastólica
Ataxia de Friedreich
Doença autossômica recessiva causada por mutações no gene FXN (doença mitocondrial)
Neurodegeneração progressiva (ataxia, fraqueza e atrofia muscular, distúrbios de linguagem, etc.)
Glicogenose (ex.: doença de Pompe)
Acúmulo de glicogênio nos lisossomos celulares, especialmente nos músculos e no coração
Frequentemente com envolvimento do sistema músculo-esquelético
Hipertensão arterial sistêmica
Hipertrofia miocárdica reativa
Geralmente simétrica, com histórico de hipertensão
Даже при наличии мутации в гене саркомерного белка клиническая выраженность и течение ГКМП зависят от дополнительных факторов:
Reguladores epigenéticos;
Hipertensão arterial concomitante;
Atividade física de alta intensidade (especialmente na adolescência);
Sexo e estado hormonal (mulheres são mais propensas a formas obstrutivas, porém costumam apresentar uma manifestação mais tardia);
Histórico familiar de morte súbita cardíaca.
Patogênese
Disfunção sarcomérica
As mutações genéticas (v. etiologia) levam a:
Aumento da sensibilidade ao cálcio;
Diminuição da eficiência contrátil;
Aumento das necessidades energéticas.
Consequências: desenvolve-se a hipertrofia compensatória do miocárdio, principalmente do septo interventricular, especialmente na região da via de saída do ventrículo esquerdo (VSVE).
Hipertrofia e alterações do relaxamento ventricular (disfunção diastólica)
O espessamento das paredes leva a:
Diminuição da complacência ventricular esquerda;
Deificuldade do enchimento diastólico;
Aumento da pressão diastólica.
Consequências: desenvolvimento de congestão pulmonar, manifestada por falta de ar e outros sintomas de insuficiência cardíaca com fração de ejeção preservada.
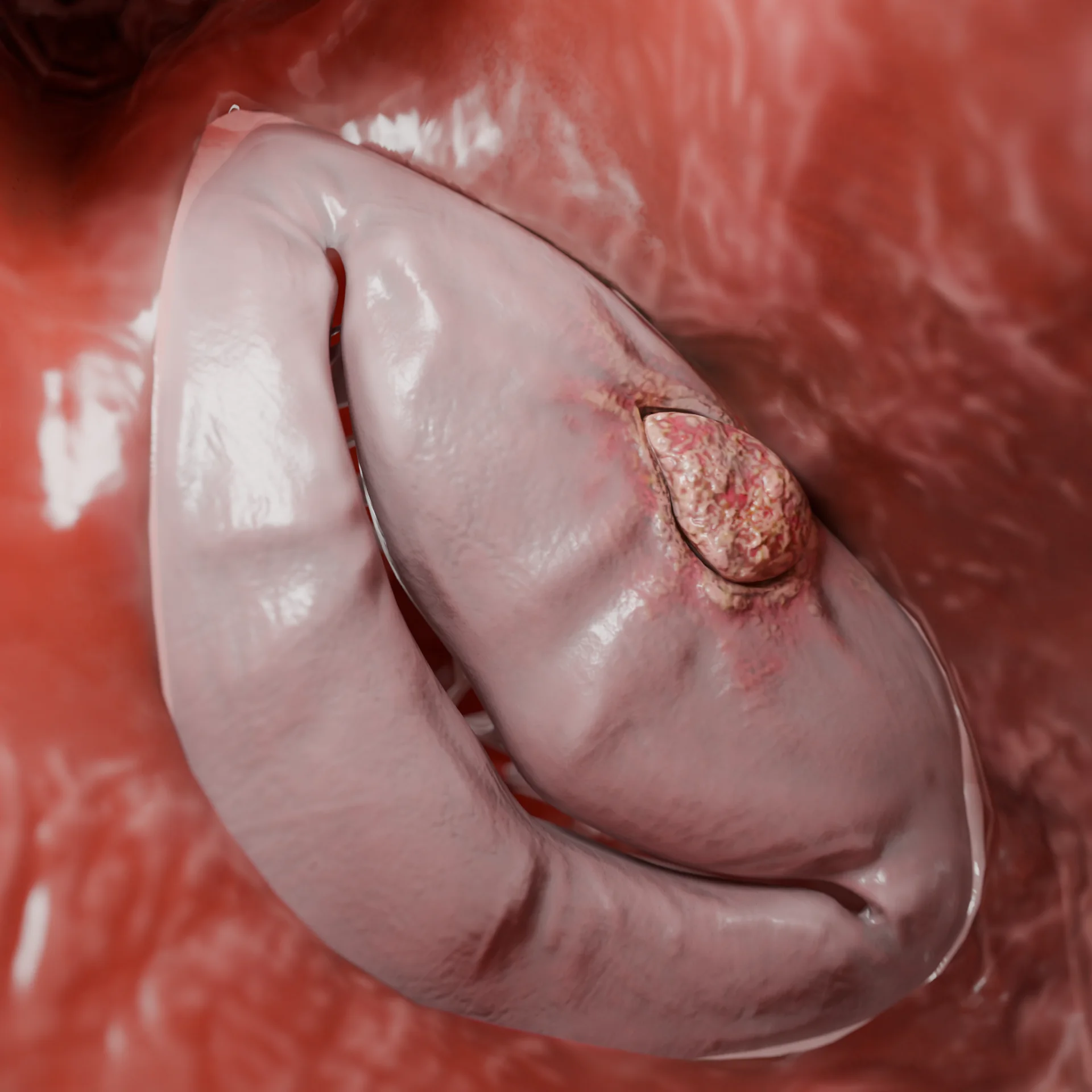
Estreitamento grave da via de saída do ventrículo esquerdo (forma obstrutiva)
Em 60–70% dos pacientes, desenvolve-se uma obstrução da via de saída do ventrículo esquerdo.
Devido ao efeito Venturi durante a sístole, ocorre: a retração da cúspide anterior da valva mitral em direção ao septo interventricular (fenômeno SAM), o agravamento do gradiente de pressão e o desenvolvimento de regurgitação mitral.
Consequências: aumento da sobrecarga hemodinâmica, agravando os sintomas e aumentando o risco de arritmias.
Obstrução da via de saída do ventrículo esquerdo devido ao espessamento do septo interventricular — Modelo 3D
Isquemia microvascular
O miocárdio hipertrofiado requer mais oxigênio, mas:
A rede capilar é incapaz de compensar o crescimento do tecido;
Há um desequilíbrio entre a demanda e a oferta de oxigênio;
Muitas vezes, observa-se a fibrose de pequenos vasos.
Consequências: desenvolve-se a isquemia miocárdica com artérias coronárias normais, levando à dor torácica, à fibrose e ao aumento do risco de arritmias.
Fibrose e instabilidade elétrica
Em resposta à isquemia e à sobrecarga mecânica do miocárdio, desenvolve-se a fibrose intersticial e focal, levando a:
Condução prejudicada dos impulsos;
Desenvolvimento de arritmias ventriculares;
Aumento do risco de morte súbita cardíaca (MSC).
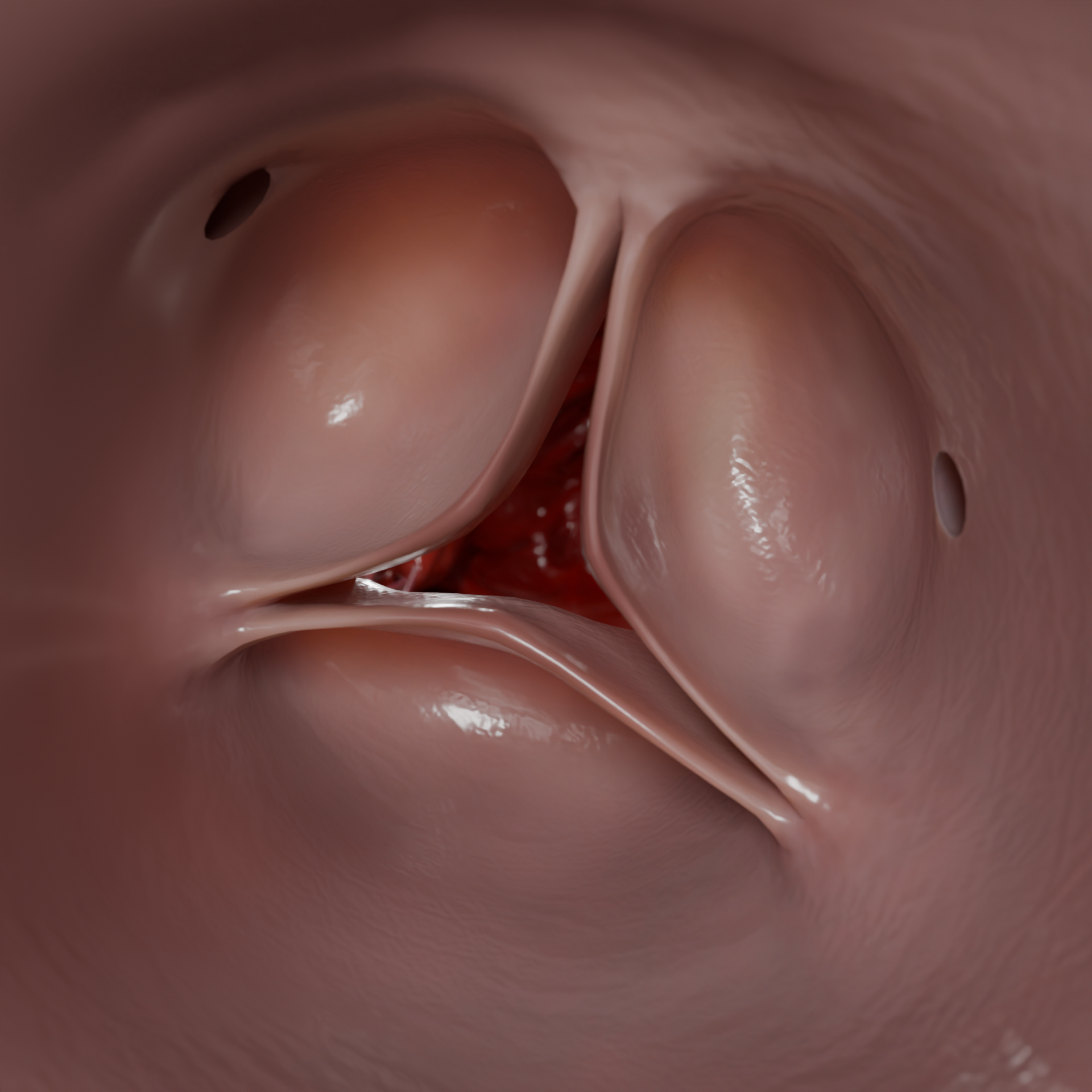
Animação 3D — cardiomiopatia hipertrófica
Manifestações clínicas
Falta de ar aos esforços, fadiga;
Dor no peito (angina) sem doença arterial coronariana;
Síncope ou pré-síncope (especialmente durante esforço físico);
Arritmias ventriculares, fibrilação atrial;
Morte súbita — especialmente em pacientes jovens e atletas que apresentam a forma obstrutiva;
Insuficiência cardíaca: pode se desenvolver tanto com fração de ejeção preservada (ICFEP) quanto com sua diminuição em estágios mais avançados. Manifesta-se como edema, ortopneia, taquicardia e diminuição da tolerância ao exercício.
Assintomática: em 25 a 30% dos pacientes, a doença é detectada por meio da análise do histórico familiar. Apresenta alto risco de complicações.
Diagnóstico de cardiomiopatia hipertrófica
A ecocardiografia (ecocardiografia transtorácica) é o método fundamental do diagnóstico inicial. Permite:
Avaliar a espessura do miocárdio. O diagnóstico é provável com uma espessura da parede ≥15 mm em adultos ou ≥13 mm em parentes de primeiro grau com diagnóstico de CMH confirmado;
Determinar a distribuição da hipertrofia: assimétrica, concêntrica, apical;
Detectar a obstrução da via de saída do ventrículo esquerdo (gradiente ≥30 mmHg, clinicamente significativo ≥50 mmHg);
Detectar o fenômeno SAM (retração sistólica da valva mitral) e a regurgitação mitral;
Avaliar a função do ventrículo esquerdo e a presença de disfunção diastólica;
Aferir o tamanho dos átrios (especialmente o átrio esquerdo, que indica risco de FA).
Ressonância magnética do coração é recomendada se:
A ecocardiografia não permitir avaliar com precisão a espessura da parede.
Houver suspeita de cardiomiopatia hipertrófica apical ou atípica.
For necessária a avaliação da fibrose miocárdica.
Revela:
Distribuição e grau de hipertrofia;
Áreas fibrosas estão associadas a um risco aumentado de arritmias e morte súbita cardíaca;
Diferenciação de fenocópias (por exemplo, amiloidose).
O eletrocardiograma (ECG) é inespecífico, mas alterações patológicas são detectadas em mais de 90% dos pacientes. Sinais de hipertrofia ventricular esquerda:
Ondas Q atípicas (nas derivações V4–V6, I, aVL) — podem mimetizar o infarto do miocárdio;
Verapamil — se os betabloqueadores forem contraindicados.
Disopiramida — como adjuvante na forma obstrutiva.
Mavacamten — um novo medicamento com efeito comprovado na redução do gradiente e na melhora dos sintomas (ESC 2023 e AHA 2020).
Tratamento cirúrgico
Indicações:
Gradiente da via de saída do ventrículo esquerdo (VSVE) ≥50 mmHg. p. em repouso ou induzido pelo exercício;
Sintomas graves (NYHA III–IV) resistentes ao tratamento com os betabloqueadores, o verapamil ou a disopiramida;
Insuficiência mitral grave associada ao fenômeno SAM.
Miectomia septal extensa:
O padrão ouro para cirurgia na cardiomiopatia hipertrófica obstrutiva;
É realizada por meio de miniesternotomia ou esternotomia completa e, em alguns casos, por meio de minitoracotomia anterior direita;
Uma seção do septo interventricular hipertrofiado é removida, eliminando o gradiente de pressão;
Se necessário, é relizado o reparo da valva mitral ou a ressecção das cordoalhas secundárias;
Possíveis complicações: obstruções, necessidade de implante de CDI, recorrência do gradiente.
A ablação septal alcoólica (intervenção endovascular) é normalmente utilizada em pacientes para os quais a cirurgia aberta é contraindicada ou apresenta riscos muito elevados.
Injeção de etanol na artéria perfurante → infarto local → redução da espessura septal;
Risco de bloqueio atrioventricular completo (até 10%) – é necessário estar preparado para o implante de marca-passo;
Resultados menos previsíveis;
Possibilidade de remoção incompleta da obstrução.
Implantação de um cardioversor-desfibrilador (CDI)
Indicações:
Morte súbita cardíaca prévia ou taquicardia ventricular sustentada;
Espessura da parede do ventrículo esquerdo é >30 mm;
Histórico familiar de morte súbita cardíaca;
Desmaio de etiologia desconhecida;
FEVE <50% com doença progressiva.
Transplante cardíaco em pacientes com insuficiência cardíaca terminal refratária, apesar do tratamento.
FAQ
1. O que é a cardiomiopatia hipertrófica?
A cardiomiopatia hipertrófica (CMH) é uma doença na qual o músculo cardíaco (geralmente a parede entre os ventrículos) se torna anormalmente espessado. Ao contrário da hipertrofia causada por hipertensão ou defeitos valvares, na CMH, o espessamento está associado às mutações genéticas em vez da sobrecarga cardíaca.
2. A CMH é hereditária? É necessário examinar parentes?
Sim, na maioria dos casos, a CMH é herdada de forma autossômica dominante. É recomendado examinar parentes de primeiro grau.
3. Quais sintomas podem indicar a CMH e quando é necessário procurar um médico?
Os sintomas incluem falta de ar, dor no peito, tontura e desmaios, especialmente durante esforço. Se você apresentar esses sintomas ou possui histórico familiar de morte súbita cardíaca, deve consultar um cardiologista.
4. A CMH é perigosa? É possível viver muitos anos com ela?
Com diagnóstico precoce e tratamento adequado, a maioria dos pacientes tem uma vida plena. No entanto, em alguns casos, a CMH aumenta o risco de arritmias e morte súbita, especialmente se não for tratada.
5. Como a CMH obstrutiva difere da CMH não obstrutiva?
Na forma obstrutiva, um septo espessado impede o fluxo sanguíneo do ventrículo esquerdo, causando sintomas mais graves. Na forma não obstrutiva, o fluxo sanguíneo não é comprometido.
6. É possível praticar atividades físicas na CMH?
Esportes intensos e competitivos não são recomendados para pessoas com CMH. O exercício moderado é permitido, mediante aprovação pelo médico.
7. Quais exames são necessários para diagnosticar a CMH?
Normalmente, são realizados o ECG, a ecocardiografia (ultrassom do coração), a ressonância magnética cardíaca, a monitorização eletrocardiográfica de 24 horas e os testes genéticos (quando indicados).
8. Como a CMH é tratada: é necessário realizar a cirurgia ou os medicamentos são suficientes?
Os medicamentos são a base do tratamento. Em casos graves, pode ser necessária a cirurgia ou a ablação septal percutânea.
9. O que é o CDI e quando ele é implantado para tratar a CMH?
O CDI é um cardioversor-desfibrilador implantável, um dispositivo que previne a morte súbita causada pelas arritmias. Ele é implantado em pacientes de alto risco por recomendação médica.
10. Há cura exata para HCM? Qual é o prognóstico?
Não há cura exata para CMH, mas seus sintomas podem ser controlados de forma eficaz. Com a abordagem correta, o prognóstico é favorável, especialmente na ausência de complicações graves.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.
São Petersburgo FL 33702, 7901 4th St N STE 300, EUA
Obrigado!
A tua mensagem foi enviada! Os nossos especialistas entrarão em contacto contigo em breve. Se tiveres mais perguntas, contacta-nos através de info@voka.io