Cardiomiopatia restritiva: etiologia, patogénese, sintomas, diagnóstico, métodos de tratamento
Kizyukevich O.Cirurgião cardiovascular, MD
12 min ler·Dezembro 23, 2025
Este artigo é apenas para fins informativos
O conteúdo deste sítio Web, incluindo texto, gráficos e outros materiais, é fornecido apenas para fins informativos. Não se destina a servir de conselho ou orientação. Relativamente ao teu estado de saúde ou tratamento específico, consulta o teu profissional de saúde.
A cardiomiopatia restritiva (CMR) é uma forma rara de cardiomiopatias primárias ou secundárias, caracterizada por comprometimento do enchimento diastólico do ventrículo esquerdo e/ou direito com função sistólica e espessura da parede normais ou quase normais. Ao mesmo tempo, o aumento da rigidez miocárdica leva ao aumento da pressão diastólica e insuficiência cardíaca congestiva.
Esta condição requer um diagnóstico diferencial complexo, uma vez que pode ser causada por uma variedade de razões, desde distúrbios genéticos a doenças sistémicas.
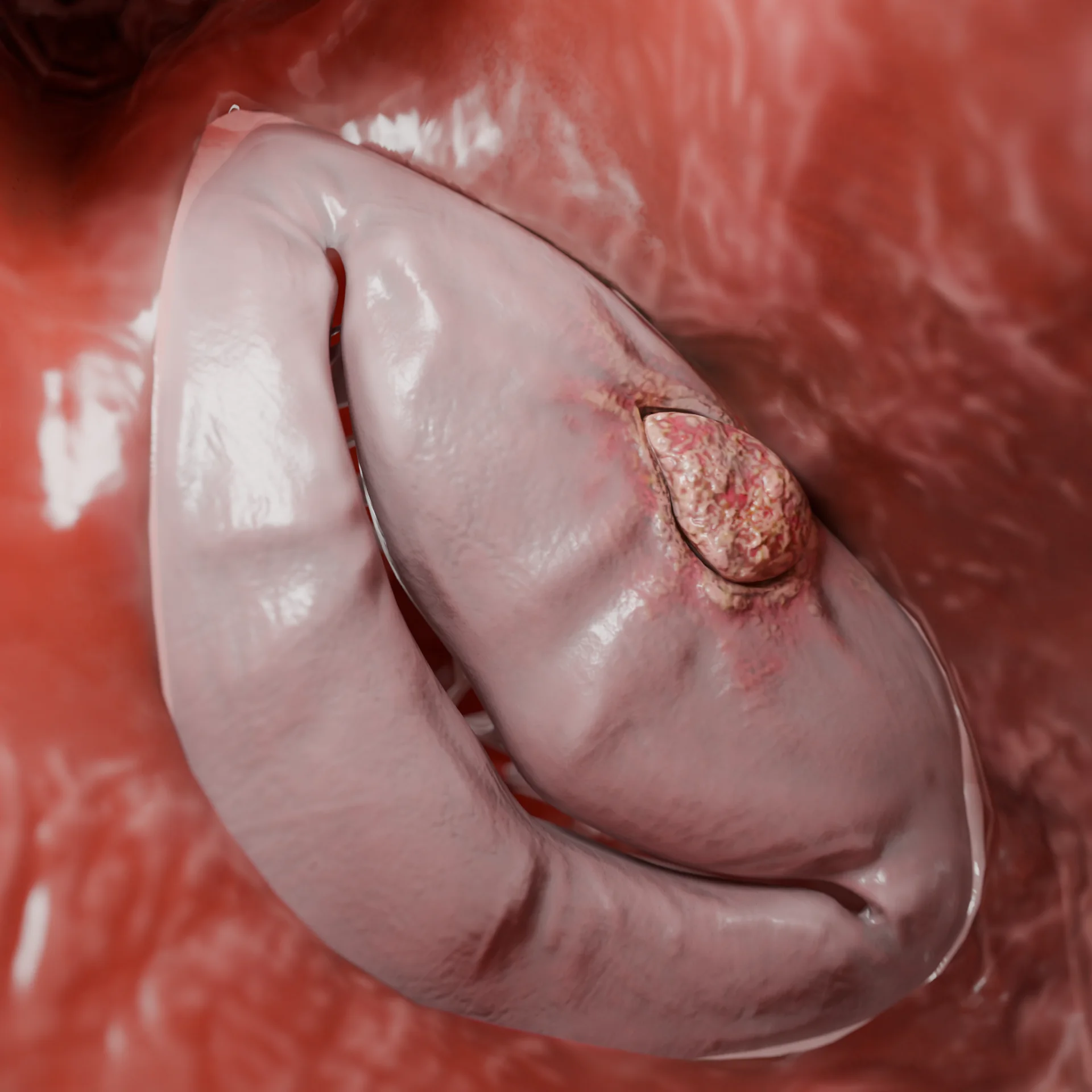
Cavidade do ventrículo esquerdo com cardiomiopatia restritiva resultante de fibrose endomiocárdica – modelo 3D
Causas da cardiomiopatia restritiva
A doença pode ser idiopática (primária) ou secundária em relação a processos sistémicos ou infiltrativos. As principais causas da cardiomiopatia restritiva podem ser divididas em vários grupos:
Doenças infiltrativas:
A amiloidose (formas AL e ATTR) é a causa mais comum nos adultos.
A sarcoidose é uma lesão granulomatosa do miocárdio, que leva à rigidez e a perturbações da condução.
Doenças de depósito:
Doença de Fabry (deficiência da α-galactosidase A).
A hemocromatose é o depósito de ferro no miocárdio, que leva à fibrose.
Lesões induzidas por radiação e quimioterapia:
Efeitos tóxicos dos antraciclinos (por exemplo, doxorrubicina).
Sequelas da radioterapia mediastínica.
Lesões endomiocárdicas:
Fibrose endomiocárdica.
A miocardiopatia restritiva de Löffler (endocardite hipereosinofílica) é uma lesão tóxica do coração por eosinófilos.
Doenças sistêmicas:
Esclerodermia, lúpus eritematoso sistémico.
Em alguns casos, a etiologia não pode ser determinada, sendo essa forma denominada idiopática.
Patogénese e classificação
O principal mecanismo de desenvolvimento da cardiomiopatia restritiva é a diminuição da complacência (elasticidade) do miocárdio com função sistólica preservada ou ligeiramente reduzida. As paredes ventriculares tornam-se rígidas e não conseguem relaxar completamente durante a diástole.
Isso desencadeia uma complexa sequência de reações patológicas:
Aumento da pressão de enchimento: As paredes rígidas dos ventrículos exigem elevada pressão de enchimento, o que conduz inevitavelmente a congestão venosa nos pulmões e na circulação sistémica.
Diminuição do débito cardíaco: Devido ao enchimento limitado, o volume sistólico e o débito cardíaco diminuem, mesmo com uma fração de ejeção normal.
Função dos átrios: A diminuição do enchimento dos ventrículos provoca um aumento compensatório da pressão auricular. Isto leva a uma dilatação atrial significativa, à diminuição da “contribuição auricular” para o enchimento ventricular e ao agravamento dos sintomas de insuficiência cardíaca.
Fibrose e remodelação: Desenvolvem-se como consequência da sobrecarga crónica de pressão, contribuindo para a progressão do fenótipo restritivo.
Arritmias:
Frequentemente desenvolvem-se fibrilação atrial, bloqueios AV, bradicardias, especialmente em casos de amiloidose ou sarcoidose, que afetam o sistema de condução. Além disso, aumenta o risco de trombose.
No final da cadeia patogenética, desenvolve-se insuficiência cardíaca diastólica refratária (não controlada) com baixo débito cardíaco, má resposta à terapia padrão e alto risco de tromboembolismo.
Características da patogénese para cada forma individual
Os mecanismos de lesão dependem da etiologia:
Na amiloidose: a deposição de proteínas entre os cardiomiócitos provoca fibrose e distúrbios de condução.
Na doença de Fabry: o acúmulo intracelular de glicosfingolipídios leva à disfunção das próprias células (disfunção miocítica).
Na sarcoidose: a inflamação granulomatosa é substituída por tecido fibroso, causando arritmias.
Classificação da cardiomiopatia restritiva
Tipos de CMR
Descrição
Primária (idiopática)
A lesão é limitada pelo miocárdio, a causa é desconhecida
Infiltrativa
Acumulação de substâncias patológicas (por exemplo, amilóide) no espaço intercelular
Doenças de depósito
Acumulação de metabolitos (ferro, glicogénio) no interior das células
Endomiocárdica
Fibrose da camada interna (por exemplo, doença de Leffler)
Provocada pela radioterapia e quimioterapia
Lesão do miocárdio devido à exposição a radiação ionizante e/ou cardiotoxicidade induzida pela quimioterapia
Manifestações clínicas
Os sintomas da cardiomiopatia restritiva estão principalmente associados à disfunção diastólica e à congestão venosa, que se manifestam da seguinte forma:
Falta de ar: inicialmente durante o esforço físico (devido ao enchimento insuficiente do ventrículo esquerdo), depois em repouso.
Ortopneia: falta de ar na posição deitada, edema pulmonar (congestão na circulação pulmonar).
Síndrome do edema: inchaço das veias jugulares, edema periférico das pernas, ascite.
Sintomas de débito cardíaco diminuído: fadiga intensa, fraqueza, hipotensão ortostática e desmaios.
Distúrbios do ritmo cardíaco: a fibrilhação auricular é uma complicação comum que agrava o estado do paciente.
Tromboembolias: o risco de acidente vascular cerebral é elevado devido à estagnação do sangue nos átrios dilatados e à arritmia.
Diagnóstico
O diagnóstico da CMR requer uma abordagem abrangente para excluir a pericardite constritiva e identificar a causa da doença.
Análises clínicas
NT-proBNP ou BNP: elevados (não específicos, marcadores da gravidade da insuficiência cardíaca).
Troponinas: podem estar moderadamente elevadas em formas infiltrativas (amiloidose), mesmo sem isquemia.
Exames de rastreio: Amilóide A sérica, eletroforese de proteínas sanguíneas, imunoglobulinas (exclusão de amiloidose AL e outras paraproteinemias), ferro e ferritina (hemochromatose), atividade GLA (doença de Fabry).
Métodos instrumentais
1. Ecocardiografia (EcoCG) Na ecocardiografia, a miocardiopatia restritiva apresenta características específicas:
espessamento das paredes com volume ventricular normal ou reduzido;
dilatação auricular, especialmente do átrio esquerdo;
rigidez do VE, enchimento diastólico restritivo (relação E/A elevada);
fração de ejeção preservada (diminui apenas nos estágios finais);
na amiloidose: um sinal específico de preservação apical «apical sparing» (redução da deformação longitudinal com preservação do segmento apical).
2. Eletrocardiografia (ECG) A CMR no ECG manifesta-se frequentemente como ondas de baixa voltagem, fibrilação atrial, bloqueios AV e arritmias ventriculares.
3. Ressonância magnética cardíaca com gadolínio (realce tardio pelo gadolínio) Padrão ouro para visualização de infiltração e fibrose:
A acumulação de contraste subendocárdico é típica da amiloidose.
A fibrose focal é típica da sarcoidose.
A ausência de acumulação específica é característica da forma idiopática.
4. Cateterismo cardíaco (hemodinâmica invasiva)
É utilizado para medir a pressão nas câmaras cardíacas. Permite diferenciar a CMR da pericardite constritiva (pela diferença de pressão nos ventrículos durante a respiração), o que é fundamental para a escolha das estratégias de tratamento.
5. Métodos complementares
Biópsia do miocárdio: O método apresenta certos riscos, mas permite verificar de forma precisa a natureza infiltrativa ou inflamatória das alterações.
Exame PYP (cintilografia 99mTc-PYP):É uma ferramenta diagnóstica bastante sensível da amiloidose ATTR. Diferencia-a da forma AL.
FDG-PET: revela inflamação ativa na sarcoidose.
TC cardíaca: para excluir calcificação do pericárdio (pericardite constritiva).
Fibrose endomiocárdica que causou o desenvolvimento de CMR – modelo 3D
Encontra mais conteúdos cientificamente exactos nas nossas redes sociais
Subscreve e não percas os recursos mais recentes
Tratamento da cardiomiopatia restritiva
As guidelines atuais dividem a terapia em sintomática e específica (direcionada à causa).
Terapia medicamentosa
Diuréticos de alça (furosemida, torasemida): tratamento sintomático da insuficiência cardíaca congestiva. Reduzem a congestão, mas requerem cautela para não diminuir o débito cardíaco.
Betabloqueadores e antagonistas do cálcio: são usados para controlar a frequência cardíaca em casos de taquicardia (com cuidado em casos de amiloidose).
Anticoagulantes: são obrigatórios em casos de fibrilação atrial para prevenir tromboses.
Terapia específica
Para amiloidose ATTR: Tafamidis.
Para a amiloidose AL: quimioterapia (bortezomib, ciclofosfamida), transplante de medula óssea.
Na doença de Fabry: terapia com chaperona farmacológica (migalastate) e terapia de reposição enzimática (agalsidase alfa/beta).
Para a sarcoidose: glicocorticóides (prednisolona) e imunossupressores (metotrexato, azatioprina).
Para a hemocromatose: quelantes de ferro (deferoxamina).
Tratamento cirúrgico
Implante de marcapasso: em casos de bradicardia grave e bloqueios AV.
Implantação de cardioversor-desfibrilhador implantável (CDI): para a prevenção da morte súbita cardíaca (especialmente após episódios de taquicardia ventricular e nas formas cicatriciais de sarcoidose).
Transplante cardíaco: indicado para insuficiência cardíaca grave refratária em doentes jovens sem lesão sistémica de órgãos.
FAQ
1. O que é a cardiomiopatia restritiva e como difere de outras formas?
A CMR é uma condição em que o miocárdio se torna rígido, perdendo a sua capacidade de relaxar. Por exemplo, ao contrário da forma dilatada, na CMR o tamanho das câmaras cardíacas geralmente não aumenta e a função sistólica permanece preservada durante um longo período.
2. Quais são os sintomas da CMR?
Falta de ar, fadiga, edema, distensão da veia jugular e ascite. Esses sintomas estão associados à estagnação do sangue nas circulações pulmonar e sistémica devido à disfunção diastólica.
3. A CMR pode ser uma doença hereditária?
Sim. Algumas formas, como a amiloidose transtiretina (ATTRv), têm natureza genética.
4. É possível curar completamente a doença?
Na maioria dos casos, a doença é progressiva. No entanto, com a identificação atempada de causas reversíveis (por exemplo, sarcoidose, miocardite eosinofílica), é possível a estabilização ou melhoria.
5. Como é diagnosticada a cardiomiopatia restritiva?
São utilizados ecocardiografia, TC/RM do coração, ECG, biomarcadores (NT-proBNP, troponinas), biópsia do miocárdio, cintilografia e testes laboratoriais para detetar amiloidose e outras causas.
6. Quais são as doenças que mais frequentemente levam à CMR?
Amiloidose (ATTR e AL), sarcoidose, hemocromatose, doenças sistémicas do tecido conjuntivo, endomiocardite eosinofílica, lesão por radiação.
7. Qual é o tratamento para a cardiomiopatia restritiva?
Tratamento da causa (por exemplo, quimioterapia para amiloidose AL), controlo dos sintomas (diuréticos, antiarrítmicos) e, em casos de insuficiência cardíaca grave, transplante cardíaco.
8. Qual é o prognóstico para a cardiomiopatia restritiva?
O prognóstico depende da etiologia. A amiloidose AL está associada a um prognóstico desfavorável, enquanto que na ATTR pode-se observar um curso mais estável com terapia direcionada.
9. É possível praticar desporto com o diagnóstico de CMR?
A atividade física deve ser limitada. Apenas exercícios físicos ligeiros sob supervisão médica são permitidos.
10. Quando é indicado o transplante cardíaco em casos de CMR?
Em insuficiência cardíaca terminal resistente à terapia, sem lesões extracardíacas e na ausência de contraindicações. Isto é especialmente relevante quando tratamentos específicos (por exemplo, amiloidose AL) são ineficazes.
Rapezzi C, Aimo A, Barison A, et al. Restrictive cardiomyopathy: definition and diagnosis. Eur Heart J. 2022 Dec 1;43(45):4679-4693. doi: 10.1093/eurheartj/ehac543.
3.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi: 10.1093/eurheartj/ehad194.
4.
Muchtar E, Blauwet LA, Gertz MA. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017 Set 15;121(7):819-837. doi: 10.1161/CIRCRESAHA.117.310982.
5.
Vio R, Angelini A, Basso C, et al. Hypertrophic Cardiomyopathy and Primary Restrictive Cardiomyopathy: Similarities, Differences and Phenocopies. J Clin Med. 2021 May 1;10(9):1954. doi: 10.3390/jcm10091954.
6.
Garcia MJ. Constrictive Pericarditis Versus Restrictive Cardiomyopathy? J Am Coll Cardiol. 2016 May 3;67(17):2061-76. doi: 10.1016/j.jacc.2016.01.076.
7.
Arbeláez-Cortés Á, Quintero-González DC, Cuesta-Astroz Y, et al. Restrictive cardiomyopathy in a patient with systemic sclerosis and Fabry disease: a case-based review. Rheumatol Int. 2020 Mar;40(3):489-497. doi: 10.1007/s00296-019-04453-y.
São Petersburgo FL 33702, 7901 4th St N STE 300, EUA
Obrigado!
A tua mensagem foi enviada! Os nossos especialistas entrarão em contacto contigo em breve. Se tiveres mais perguntas, contacta-nos através de info@voka.io