Cardiomyopathie restrictive : étiologie, pathogénie, symptômes, diagnostic, méthodes de traitement
Kizyukevich O.Chirurgien cardiovasculaire, MD
12 minutes de lecture·décembre 23, 2025
Cet article est uniquement destiné à des fins d'information
Le contenu de ce site web, y compris les textes, les graphiques et autres matériels, est fourni à titre d'information uniquement. Il ne s'agit pas d'un avis ou d'un conseil. En ce qui concerne votre état de santé ou votre traitement spécifique, veuillez consulter votre fournisseur de soins de santé.
La cardiomyopathie restrictive (CMR) est une forme rare de cardiomyopathie primaire ou secondaire, caractérisée par un trouble du remplissage diastolique du ventricule gauche et/ou droit, avec une fonction systolique normale ou subnormale et une épaisseur pariétale conservée. Elle se traduit par une rigidité myocardique marquée, entraînant une élévation des pressions diastoliques et une insuffisance cardiaque congestive.
Ce tableau nécessite un diagnostic différentiel complexe, car il peut être secondaire à de multiples étiologies, allant des anomalies génétiques aux maladies systémiques.
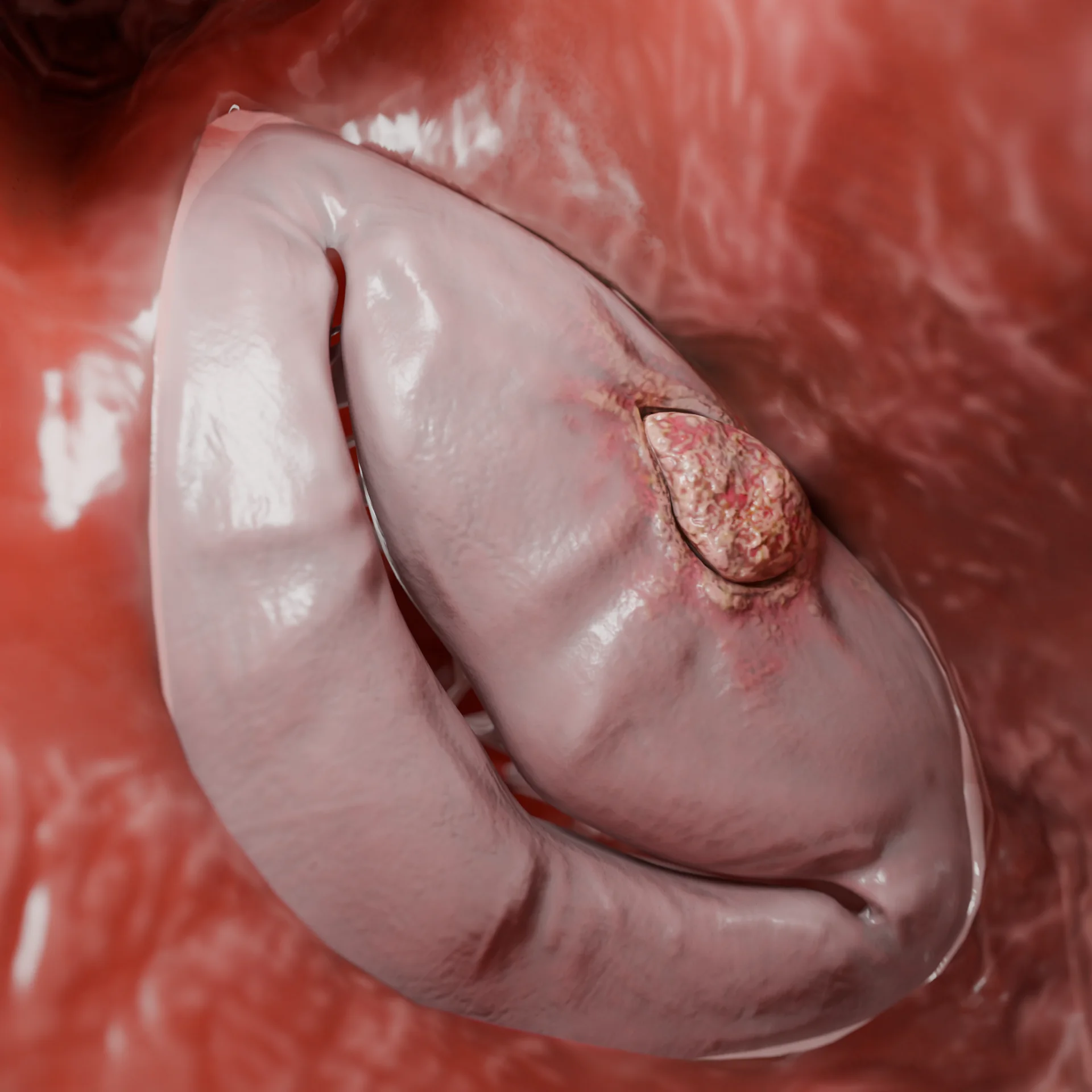
Cavité du ventricule gauche dans la cardiomyopathie restrictive secondaire à un fibroélastose endomyocardique – modèle 3D
Causes de la cardiomyopathie restrictive
La maladie peut être idiopathique (primitive) ou secondaire à des processus systémiques ou infiltratifs. Les principales causes se répartissent en plusieurs groupes :
Maladies infiltratives :
Amylose (formes AL et ATTR) – cause la plus fréquente chez l’adulte.
Sarcoïdose – atteinte granulomateuse du myocarde entraînant rigidité et troubles de conduction.
Maladies de surcharge :
Maladie de Fabry (déficit en α-galactosidase A).
Hémochromatose – dépôts de fer dans le myocarde aboutissant à une fibrose.
Lésions radio- ou chimio-induites :
Toxicité des anthracyclines (ex. : doxorubicine).
Séquelles d’irradiation médiastinale.
Atteintes endomyocardiques :
Fibrose endomyocardique.
Cardiomyopathie restrictive de Löffler (endocardite hyperéosinophilique) — lésion toxique cardiaque par les éosinophiles.
Dans certains cas, l’étiologie reste inconnue : forme idiopathique.
Pathogénie et classification
Le mécanisme principal est la diminution de la compliance (distensibilité) myocardique avec conservation ou légère altération de la fonction systolique. Les parois ventriculaires deviennent rigides et ne peuvent plus se relâcher correctement en diastole.
Cela déclenche une cascade pathologique :
Élévation des pressions de remplissage : les parois rigides nécessitent une pression élevée pour le remplissage → congestion veineuse pulmonaire et systémique.
Réduction du débit cardiaque : volume d’éjection systolique et débit-minute diminués malgré une fraction d’éjection souvent conservée.
Rôle des oreillettes : dilatation compensatrice des oreillettes, perte de la contribution auriculaire au remplissage ventriculaire. Cela entraîne leur dilatation marquée dilatation, une réduction de la contribution auriculaire au remplissage ventriculaire et une aggravation des symptômes d’insuffisance cardiaque.
Fibrose et remodelage : conséquence de la surcharge chronique en pression, favorisant la progression du phénotype restrictif.
Arythmies : fibrillation auriculaire fréquente, blocs auriculo-ventriculaires, bradycardies (surtout amylose et sarcoïdose). Et augmente également le risque de thrombogenèse.
À l’issue de la chaîne pathogénique se développe une insuffisance cardiaque diastolique réfractaire à bas débit cardiaque, peu répondante au traitement standard et associée à un risque élevé d’événements thromboemboliques.
Particularités pathogéniques selon l’étiologie
Les mécanismes lésionnels varient selon l’étiologie :
Amylose : dépôts extracellulaires d’amyloïde → fibrose et troubles de conduction.
Maladie de Fabry : accumulation intracellulaire de glycosphingolipides → dysfonction myocytaire directe.
Sarcoïdose : remplacement du granulome par fibrose → arythmies.
Classification de la cardiomyopathie restrictive
Type
Description
Primitive (idiopathique)
Atteinte limitée au myocarde, cause inconnue
Infiltrative
Accumulation interstitielle de substances pathologiques (ex. : amyloïde)
Maladies de surcharge
Accumulation intracellulaire de métabolites (fer, glycogène…)
Endomyocardique
Fibrose de l’endocarde (ex. : maladie de Löffler)
Radio- et chimio-induite
Lésions secondaires à irradiation et/ou agents cardiotoxiques
Manifestations cliniques
Les symptômes sont principalement liés à la dysfonction diastolique et à la congestion veineuse :
Dyspnée : d’effort puis de repos.
Orthopnée, œdème aigu du poumon (congestion pulmonaire).
Syndrome œdémateux :turgescence jugulaire, œdèmes des membres inférieurs, ascite.
Signes de bas débit : fatigue extrême, faiblesse, hypotension orthostatique, syncopes.
Troubles du rythme : fibrillation auriculaire fréquente, aggravant l’hémodynamique.
Thromboembolies : risque élevé d’AVC par stase dans les oreillettes dilatées.
Diagnostic
Le diagnostic exige une approche multimodale pour exclure le péricardite constrictive et identifier la cause.
Examens biologiques
NT-proBNP / BNP : élevés (marqueurs de gravité).
Troponines : discrètement élevées dans les formes infiltratives sans ischémie.
Bilan étiologique : protéine amyloïde A sérique, électrophorèse des protéines immunoglobulines (amylose AL), ferritinémie et saturation de la transferrine (hémochromatose), activité α-galactosidase A (Fabry).
Méthodes instrumentales
1. Échocardiographie À l’échocardiographie, la cardiomyopathie restrictive présente des signes caractéristiques :
Épaississement pariétal avec cavités ventriculaires de taille normale ou réduite.
Dilatation bi-auriculaire marquée.
Remplissage diastolique de type restrictif (rapport E/A élevé, E/e’ élevé).
Permet la mesure directe des pressions intracardiaques. Il est essentiel pour différencier la cardiomyopathie restrictive du péricardite constrictive (par l’absence de variation respiratoire significative des pressions intraventriculaires et l’absence d’égalisation des pressions diastoliques), ce qui est déterminant pour le choix de la stratégie thérapeutique.
5. Méthodes complémentaires
Biopsie endomyocardique : procédure associée à certains risques, mais permet une vérification histologique précise du caractère infiltratif ou inflammatoire des lésions.
Scintigraphie osseuse Tc-99m-PYP : méthode très sensible pour le diagnostic de l’amylose cardiaque de type ATTR ; permet de la différencier de la forme AL.
TEP au FDG : met en évidence l’inflammation active dans la sarcoïdose cardiaque.
Scanner cardiaque : indiqué pour exclure une calcification péricardique (péricardite constrictive).
Fibrose endomyocardique responsable d’une cardiomyopathie restrictive – modèle 3D
Retrouvez d’autres contenus scientifiquement exacts sur nos médias sociaux
Abonnez-vous et ne manquez pas les dernières ressources
Traitement de la cardiomyopathie restrictive
La prise en charge associe un traitement symptomatique et un traitement étiologique spécifique.
Traitement médicamenteux
Diurétiques de l’anse (furosémide, torasémide) : traitement symptomatique de l’insuffisance cardiaque congestive Réduisent la congestion, mais à utiliser avec prudence afin de ne pas diminuer excessivement le débit cardiaque.
Bêta-bloquants et antagonistes calciques non dihydropyridiniques : indiqués pour le contrôle de la fréquence cardiaque en cas de tachycardie (avec grande prudence dans l’amylose cardiaque).
Anticoagulants oraux : obligatoires en cas de fibrillation auriculaire pour la prévention des événements thromboemboliques.
Traitement étiologique spécifique
Amylose ATTR : tafamidis.
Amylose AL : chimiothérapie (bortézomib, cyclophosphamide), transplantation de moelle osseuse.
Maladie de Fabry : thérapie chaperonne (migalastat), thérapie de remplacement enzymatique (agalsidase alfa/bêta).
Sarcoïdose : glucocorticoïdes (prednisolone) et immunosuppresseurs (méthotrexate, azathioprine).
Hémochromatose : chélateurs du fer (déféroxamine).
Thérapie chirurgicale
Implantation d’un stimulateur cardiaque : indiquée en cas de bradycardie sévère et de blocs auriculo-ventriculaires.
Implantation d’un défibrillateur automatique implantable (DAI) : prévention de la mort subite cardiaque (en particulier après épisodes de tachycardie ventriculaire documentés et dans les formes cicatricielles de sarcoïdose).
Transplantation cardiaque : indiquée en cas d’insuffisance cardiaque sévère réfractaire chez les patients jeunes sans atteinte systémique extracardiaque majeure.
FAQ
1. Qu’est-ce que la cardiomyopathie restrictive et en quoi diffère-t-elle des autres formes ?
La cardiomyopathie restrictive est une maladie dans laquelle le myocarde devient rigide et perd sa capacité de relaxation. Par exemple, contrairement à la forme dilatée, les cavités cardiaques ne sont généralement pas augmentées de volume et la fonction systolique reste longtemps conservée.
2. Quels sont les symptômes caractéristiques de la cardiomyopathie restrictive ?
Dyspnée, fatigue rapide, œdèmes, turgescence des veines jugulaires, ascite. Ils sont liés à la congestion veineuse dans les circulations pulmonaire et systémique secondaire à la dysfonction diastolique.
3. La cardiomyopathie restrictive peut-elle être héréditaire ?
Oui. Oui. Certaines formes, par exemple l’amylose à transthyrétine de variante génétique (ATTRv), sont d’origine héréditaire.
4. Peut-on guérir complètement la maladie ?
Dans la majorité des cas, la maladie est progressive. Cependant, en cas de détection précoce d’une cause réversible (sarcoïdose, myocardite hyperéosinophilique par exemple), une stabilisation ou une amélioration est possible.
5. Comment diagnostique-t-on la cardiomyopathie restrictive ?
Par échocardiographie, TDM/IRM cardiaque, ECG, biomarqueurs (NT-proBNP, troponines), biopsie endomyocardique, scintigraphie et bilan biologique ciblé pour identifier l’amylose et les autres étiologies.
Quelles maladies sont le plus souvent à l’origine d’une cardiomyopathie restrictive ?
Amylose (ATTR et AL), sarcoïdose, hémochromatose, maladies systémiques du tissu conjonctif, endocardite hyperéosinophilique, séquelles d’irradiation.
7. Quel traitement est appliqué dans la cardiomyopathie restrictive ?
Traitement de la cause (ex. : chimiothérapie dans l’amylose AL), contrôle symptomatique (diurétiques, antiarythmiques), transplantation cardiaque en cas d’insuffisance cardiaque sévère.
8. Quel est le pronostic de la cardiomyopathie restrictive ?
Le pronostic dépend de l’étiologie. L’amylose AL est associée à un pronostic défavorable, tandis que l’ATTR peut avoir une évolution plus stable sous traitement ciblé.
9. Peut-on pratiquer un sport avec un diagnostic de la cardiomyopathie restrictive ?
L’activité physique doit être limitée. Seules des charges légères sous contrôle médical sont autorisées.
10. Quand la transplantation cardiaque est-elle indiquée dans la cardiomyopathie restrictive ?
En cas d’insuffisance cardiaque terminale réfractaire au traitement, sans atteinte extracardiaque systémique et en l’absence de contre-indications. Elle est particulièrement pertinente lorsque le traitement spécifique est inefficace (ex. : amylose AL).
Références
1.
Catalogue VOKA. [Ressource électronique.
https://catalog.voka.io/
2.
Rapezzi C, Aimo A, Barison A, et al. Restrictive cardiomyopathy: definition and diagnosis. [Cardiomyopathie restrictive : définition et diagnostic] Eur Heart J. 2022 Dec 1;43(45):4679-4693. doi: 10.1093/eurheartj/ehac543.
3.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies (Lignes directrices de l’ESC pour la prise en charge des cardiomyopathies). Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi : 10.1093/eurheartj/ehad194.
Vio R, Angelini A, Basso C, et al. Hypertrophic Cardiomyopathy and Primary Restrictive Cardiomyopathy: Similarities, Differences and Phenocopies. [Cardiomyopathie hypertrophique et cardiomyopathie restrictive primitive : similitudes, différences et phénocopies] J Clin Med. 2021 May 1;10(9):1954. doi: 10.3390/jcm10091954.
6.
Garcia MJ. Constrictive Pericarditis Versus Restrictive Cardiomyopathy? [Péricardite constrictive versus cardiomyopathie restrictive ?] J Am Coll Cardiol. 2016 May 3;67(17):2061-76. doi : 10.1016/j.jacc.2016.01.076.
7.
Arbeláez-Cortés Á, Quintero-González DC, Cuesta-Astroz Y, et al. Restrictive cardiomyopathy in a patient with systemic sclerosis and Fabry disease: a case-based review. [Cardiomyopathie restrictive chez un patient atteint de sclérodermie systémique et de maladie de Fabry : revue basée sur un cas clinique avec revue de la littérature] Rheumatol Int. 2020 Mar;40(3):489-497. doi: 10.1007/s00296-019-04453-y.
St. Petersburg FL 33702, 7901 4th St N STE 300, États-Unis
Merci de votre attention !
Votre message est envoyé ! Nos experts vous contacteront dans les plus brefs délais. Si vous avez d'autres questions, n'hésitez pas à nous contacter à l'adresse suivante : info@voka.io