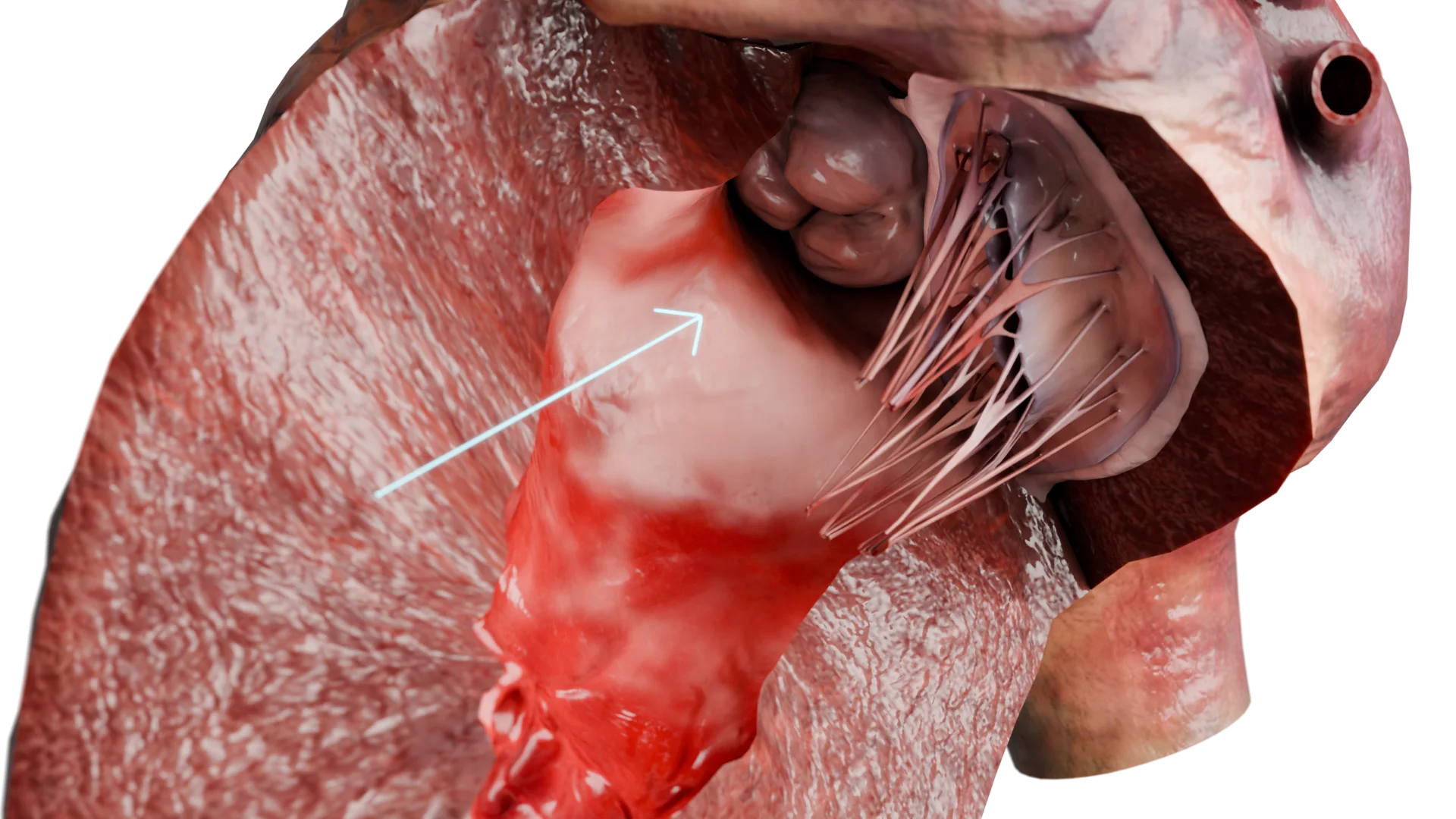
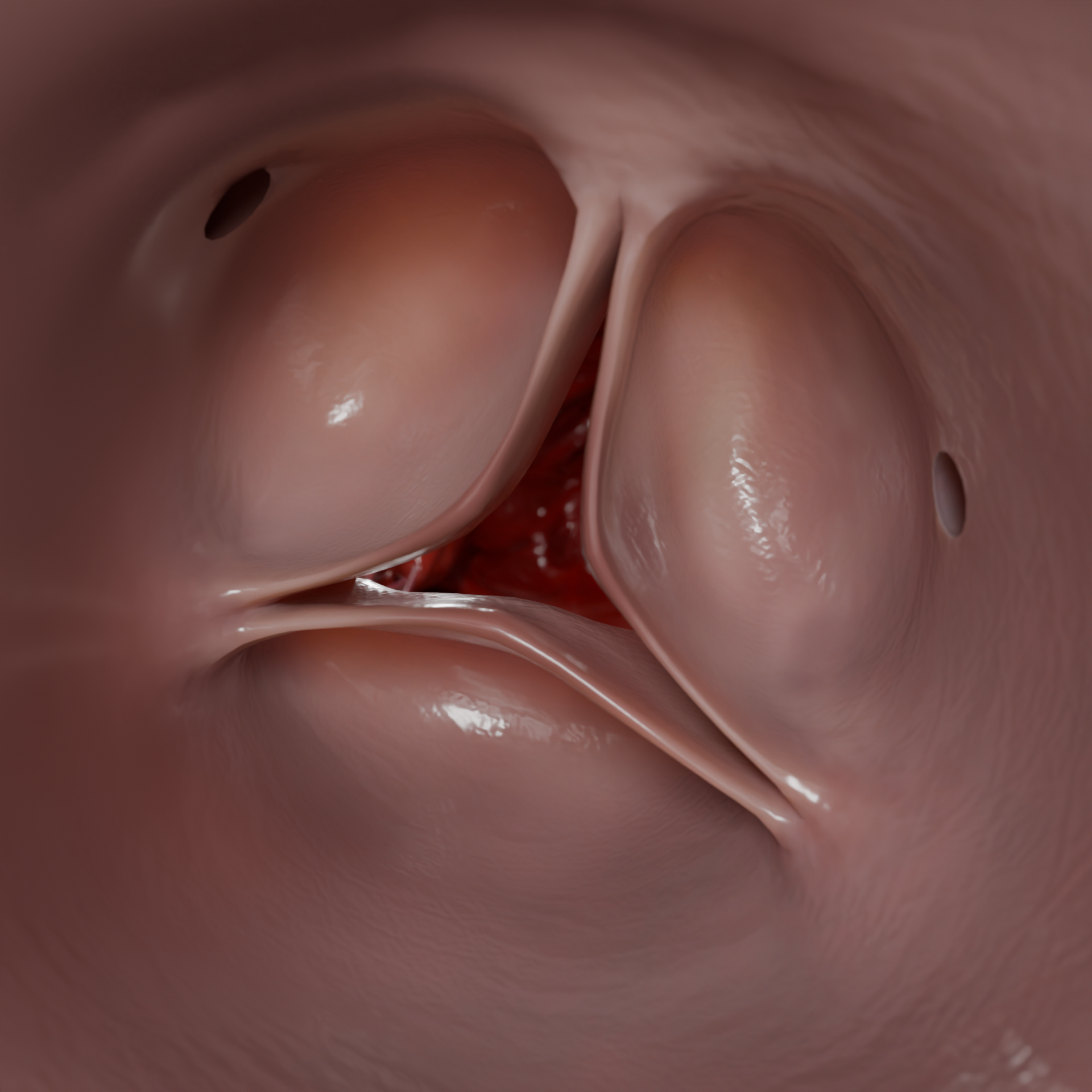
La miocardiopatía hipertrófica (MCH) es una enfermedad miocárdica primaria caracterizada por una hipertrofia inexplicable de la pared ventricular izquierda, casi siempre asimétrica, sin agrandamiento de la cavidad. La enfermedad se basa en mutaciones de genes que codifican proteínas sarcoméricas, lo que provoca anomalías morfológicas, eléctricas y hemodinámicas.Engrosamiento de la pared ventricular izquierda en HCMP – Modelo 3D
La HCMD es una de las formas hereditarias más frecuentes de miocardiopatía y se da en aproximadamente 1 de cada 500 adultos. Afecta más a los hombres que a las mujeres (proporción aproximada 3:2), pero a las mujeres se les suele diagnosticar a una edad más tardía y padecen una enfermedad más grave.
Etiología
La enfermedad se basa en una alteración de la síntesis y la función de las proteínas contráctiles sarcoméricas, pero en algunos casos se han identificado formas secundarias asociadas a trastornos sistémicos y metabólicos.
Hasta el 60-70% de los casos de miocardiopatía hipertrófica son de naturaleza genética monomutante.
Genes asociados al desarrollo de la HCMD
Gene
Proteína
Frecuencia de mutación
MYH7
miosina β-cadena pesada
MYH7 y MYBPC3 representan alrededor del 70% de los casos de mutación
MYBPC3
proteína C de unión a la miosina
MYH7 y MYBPC3 representan alrededor del 70% de los casos de mutación
TNNT2
Troponina T
Alrededor del 5
TNNI3
Troponina I
<5%
TPM1
Tropomiosina
<5%
La enfermedad se transmite de forma autosómica dominante, es decir, basta con un solo gen alterado de uno de los progenitores. En este caso, la probabilidad de la enfermedad es alta, pero la gravedad y la forma de las manifestaciones pueden variar mucho incluso dentro de la misma familia.
En aproximadamente el 30% de los casos, la mutación se produce por primera vez, sin antecedentes familiares.
Algunas enfermedades pueden imitar el cuadro clínico de la miocardiopatía hipertrófica, pero tienen un origen patogenético diferente. Es muy importante distinguirlas de la forma primaria (sarcomérica), ya que el tratamiento y el pronóstico son diferentes.
Variantes con hipertrofia secundaria (fenotipos HCMP)
Enfermedad
Mecanismo
Características
Enfermedad de Fabry
Enfermedad hereditaria por almacenamiento lisosómico (deficiencia de la enzima α-galactosidasa A)
Signos de afectación sistémica (angioqueratomas, neuropatía, proteinuria)
Amiloidosis del corazón
Depósito de proteínas (AL, ATTR) en el miocardio
Engrosamiento de la pared, contractilidad reducida, disfunción diastólica
Ataxia de Friedreich
Enfermedad autosómica recesiva causada por mutaciones en el gen FXN (enfermedad mitocondrial)
Neurodegeneración progresiva (ataxia, debilidad y atrofia muscular, alteraciones del habla, etc.).
Glucogenosis (por ejemplo, enfermedad de Pompe)
Acumulación de glucógeno en los lisosomas de las células, especialmente en el músculo y el corazón
A menudo con afectación del músculo esquelético
Hipertensión arterial sistémica
Hipertrofia miocárdica reactiva
Típicamente simétrico, con antecedentes de hipertensión
Incluso en presencia de una mutación en el gen de la proteína sarcomérica, la gravedad clínica y el curso de la PGCH dependen de factores adicionales:
Reguladores epigenéticos;
Hipertensión arterial asociada;
Actividad física de alta intensidad (especialmente durante la adolescencia);
El sexo y el estado hormonal (las mujeres tienen más probabilidades de presentar formas obstructivas pero de manifestación más tardía);
Antecedentes familiares de muerte súbita cardiaca.
Patogénesis
Trastorno de la función sarcomérica
Las mutaciones en los genes (ver etiología) provocan:
Hipersensibilidad al calcio;
Reducción de la eficacia contráctil;
Aumento de las necesidades energéticas.
Consecuencia: se desarrolla una hipertrofia miocárdica compensatoria, predominantemente del tabique interventricular, especialmente en el tracto de salida del ventrículo izquierdo (TSVI).
Hipertrofia y alteración de la relajación (disfunción diastólica)
El engrosamiento de la pared conduce a:
Distensibilidad ventricular izquierda reducida;
Alteración de su llenado en diástole;
Aumento de la presión diastólica.
Consecuencia: desarrollo de estasis en el pequeño círculo de circulación sanguínea, que se manifiesta por disnea y otros síntomas de insuficiencia cardiaca con fracción de eyección preservada.
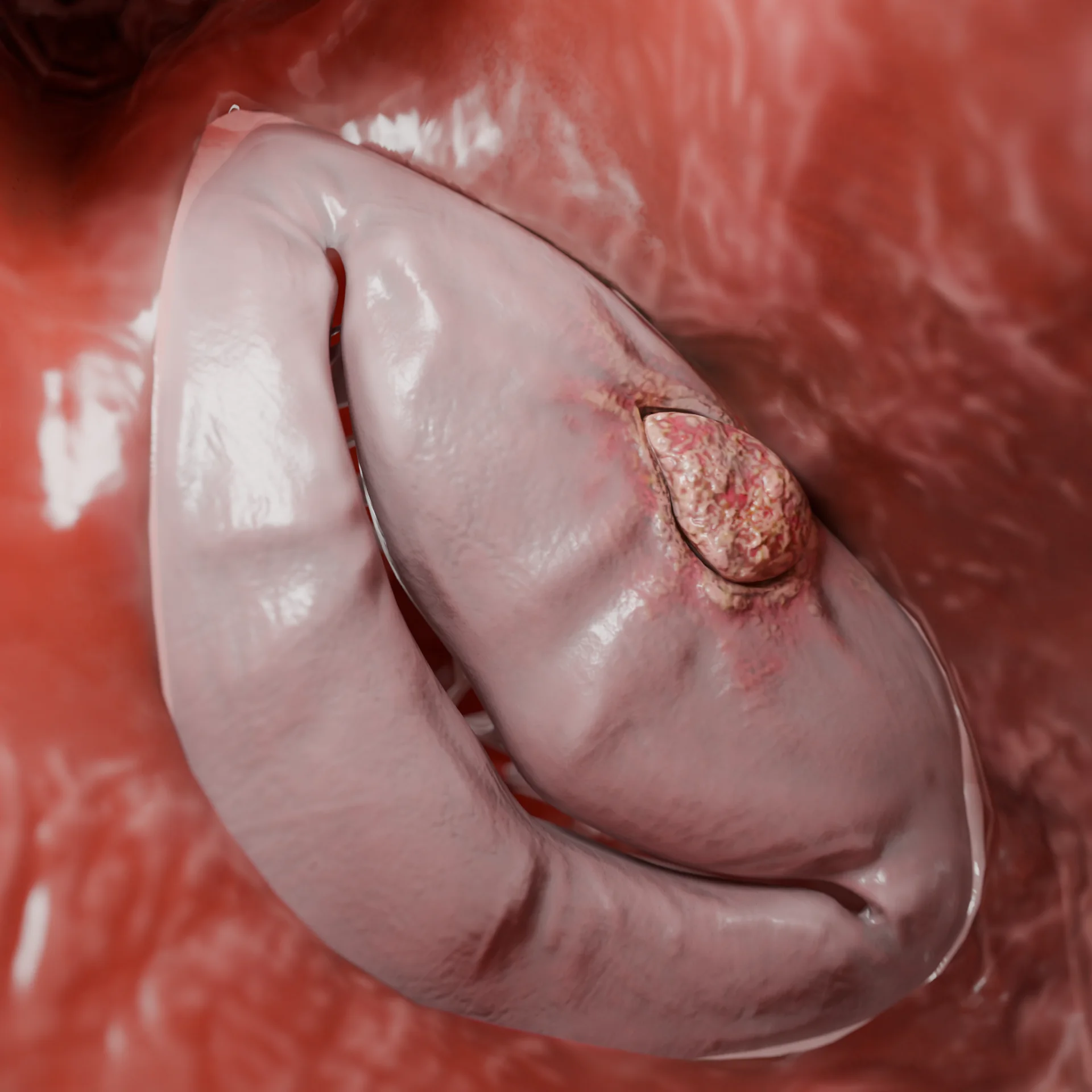
Estrechamiento grave del tracto de salida del VI (forma obstructiva)
En el 60-70% de los pacientes se forma una obstrucción de la VTE.
Debido al efecto Venturi durante la sístole, se produce: retracción de la valva anterior de la válvula mitral hacia el tabique (fenómeno SAM), agravamiento del gradiente de presión, desarrollo de regurgitación mitral.
Consecuencia: aumenta la sobrecarga hemodinámica, lo que exacerba los síntomas y aumenta el riesgo de arritmias.
Obstrucción de la VTE por engrosamiento del tabique interventricular – Modelo 3D
Isquemia microvascular
El miocardio hipertrofiado necesita más oxígeno, pero:
La red capilar no tiene tiempo de compensar el crecimiento del tejido;
Hay un desequilibrio entre la demanda y el suministro de oxígeno;
A menudo se detecta fibrosis de vasos de pequeño calibre.
Consecuencia: se produce isquemia miocárdica en las arterias coronarias normales, lo que provoca dolor torácico, fibrosis y mayor riesgo de arritmias.
Fibrosis e inestabilidad eléctrica
En respuesta a la isquemia miocárdica y a la sobrecarga mecánica, se forma fibrosis intersticial y focal, que da lugar a:
Trastorno de la conducción del impulso;
El desarrollo de arritmias ventriculares;
Aumentar el riesgo de muerte súbita cardiaca (MSC).
Animación 3D – Miocardiopatía hipertrófica
Manifestaciones clínicas
Disnea de esfuerzo, fatiga;
Dolor torácico (angina de pecho) sin enfermedad coronaria;
Síncope o presíncope (sobre todo con el ejercicio);
Arritmias ventriculares, fibrilación auricular;
VSS – especialmente en pacientes jóvenes y deportistas con forma obstructiva;
Insuficiencia cardíaca: puede presentarse con fracción de eyección preservada (IC-FEp) o con su reducción en fases tardías. Se manifiesta por edemas, ortopnea, taquicardia, disminución de la tolerancia al ejercicio;
Curso asintomático: en el 25-30% de los pacientes, la enfermedad se detecta mediante el cribado de antecedentes familiares. No excluye un alto riesgo de complicaciones.
Diagnóstico de la miocardiopatía hipertrófica
La ecocardiografía (ecocardiografía transtorácica) es un método clave del diagnóstico primario. Permite:
Evalúa el grosor del miocardio. El diagnóstico es probable cuando el grosor de la pared es ≥15 mm en adultos o ≥13 mm en familiares de primera línea con GMPc confirmado;
Define la distribución de la hipertrofia: asimétrica, concéntrica, apical;
Detectar obstrucción del tracto de salida del VI (gradiente ≥30 mmHg, clínicamente significativo ≥50 mmHg);
Detecta el fenómeno SAM (retracción sistólica de la valva mitral) y la regurgitación mitral;
Evaluar la función del VI y la presencia de disfunción diastólica;
Mide el tamaño de las aurículas (especialmente la aurícula izquierda – riesgo de FP).
La resonancia magnética cardiaca se recomienda si
La EcoCG no evalúa con precisión el grosor de la pared;
Se sospecha una forma apical o atípica de PGCH;
Es necesario evaluar la fibrosis miocárdica.
Revela:
Distribución y grado de hipertrofia;
Zonas de fibrosis: asociadas a un mayor riesgo de arritmias y EVS;
Diferenciación con fenocopias (por ejemplo, amiloidosis).
ECG (electrocardiograma): inespecífico, pero se detectan cambios patológicos en más del 90% de los pacientes. Signos de hipertrofia ventricular izquierda:
Dientes Q atípicos (en derivaciones V4-V6, I, aVL) – pueden simular un infarto de miocardio;
Cambios en el segmento ST e inversión de la onda T;
Trastornos del ritmo: fibrilación auricular, extrasístoles ventriculares, taquicardia ventricular;
Bloqueo auriculoventricular o intraventricular.
Monitorización Holter. Indicaciones:
Sospecha de arritmias (VE, VT, FP);
Síncope o presíncope;
Evaluación de la gravedad de las alteraciones del ritmo e indicaciones de DAI.
Una prueba de esfuerzo (cinta rodante o cicloergometría). Detenido por:
Evaluaciones de la tolerancia al ejercicio;
Detección de obstrucción VTE inducible;
Determinaciones de gradiente frente a la carga;
Identificación de síntomas coronarios sin estenosis de las arterias coronarias.
Pruebas genéticas. Recomendado:
Pacientes con GMPc confirmado (especialmente pacientes jóvenes o con antecedentes familiares de CHC);
Para examinar a familiares de primera línea (hijos, hermanos, padres);
En las sospechas de fenocopias (enfermedad de Fabry, enfermedades mitocondriales, etc.).
Genes identificados: MYH7, MYBPC3, TNNT2, TNNI3, TPM1 son los genes más frecuentes.
Marcadores de laboratorio:
NT-proBNP / BNP – elevado en sobrecarga de presión, disfunción diastólica;
Troponina T/I: puede estar moderadamente elevada en la isquemia microvascular.
Encuentra más contenido científicamente preciso en nuestras redes sociales
Suscríbete y no te pierdas los últimos recursos
Tratamiento de la miocardiopatía hipertrófica
Modificación del estilo de vida:
Exclusión de actividad física intensa y deportes;
Control de la tensión arterial y del peso corporal;
Verapamilo – en caso de contraindicación a los β-bloqueantes;
Disopiramida – como coadyuvante en la forma obstructiva;
Mavacamten es un nuevo fármaco con un efecto demostrado de reducción del gradiente y mejora de los síntomas (según ESC 2023 y AHA 2020).
Tratamiento quirúrgico
Indicaciones:
Gradiente del tracto de salida del ventrículo izquierdo (LVOG) ≥50 mmHg en reposo o en provocación;
Síntomas graves (NYHA III-IV) no susceptibles de tratamiento con β-bloqueantes, verapamilo o disopiramida;
Regurgitación mitral prominente asociada al fenómeno SAM.
Miectomía septal ampliada:
El estándar de oro de la cirugía para la CGMP obstructiva;
Se realiza a través de una mininotomía o una esternotomía completa, en algunos casos posiblemente a través de una minitoracotomía anterior derecha;
Se extirpa una sección del tabique interventricular hipertrofiado, eliminando el gradiente de presión;
Si es necesario, se realiza una plastia de la válvula mitral o una resección de las cuerdas secundarias;
Posibles complicaciones: obstrucciones, necesidad de DAI, reaparición del gradiente.
La ablación septal con alcohol (intervención endovascular) se utiliza más comúnmente en pacientes para los que la cirugía abierta está contraindicada o tiene riesgos demasiado elevados.
Inyección de etanol en la arteria perforante → infarto local → reducción del grosor septal;
Riesgo de bloqueo AV completo (hasta un 10%) – se requiere preparación para la implantación de un SCE;
Menos previsibilidad de los resultados;
Posibilidad de eliminación incompleta de la obstrucción.
Implantación de un desfibrilador cardioversor (DAI)
Indicaciones:
VSS experimentado o VT sostenido;
Espesor de la pared del VI >30 mm;
Historia Familiar VSS;
Síncope de etiología poco clara;
FLM <50% en el curso progresivo.
Trasplante cardíaco en pacientes con CH terminal refractaria a pesar del tratamiento.
FAQ
1. ¿Qué es la miocardiopatía hipertrófica?
La miocardiopatía hipertrófica (MCH) es una enfermedad en la que el músculo cardiaco (normalmente el tabique entre los ventrículos) se engrosa de forma anormal. A diferencia de la hipertrofia debida a la hipertensión o a defectos valvulares, en la MCHF el engrosamiento se debe a mutaciones genéticas y no a una sobrecarga del corazón.
2. ¿La cGMP es hereditaria? ¿Es necesario examinar a los familiares?
Sí, en la mayoría de los casos, la GMPc se hereda con un patrón autosómico dominante. Se recomienda examinar a los familiares de primera línea.
3. ¿Qué síntomas pueden indicar cGMP y cuándo debo buscar atención médica?
Los síntomas incluyen dificultad para respirar, dolor torácico, mareos, desmayos, sobre todo al hacer esfuerzos. Si tienes estas molestias o antecedentes familiares de muerte súbita, debes consultar a un cardiólogo.
4 ¿Es peligroso el cGMP? ¿Es posible vivir con ello durante mucho tiempo?
Con un diagnóstico a tiempo y un tratamiento adecuado, la mayoría de los pacientes viven una vida plena. Sin embargo, en algunos casos, la HCMR aumenta el riesgo de arritmias y muerte súbita, sobre todo sin tratamiento.
5. ¿En qué se diferencia el GMPc obstructivo del GMPc no obstructivo?
En la forma obstructiva, el tabique engrosado interfiere en el flujo de salida de la sangre del ventrículo izquierdo, lo que provoca síntomas más graves. En la forma no obstructiva, el flujo de salida no se ve afectado.
6. ¿Puedo hacer ejercicio cuando tengo un cGMP?
En cGMP no se recomiendan los deportes intensos y competitivos. Se permite una actividad física moderada, acordada con el médico tratante.
7. ¿Qué exámenes son necesarios para hacer un diagnóstico de cGMP?
Normalmente se realiza un ECG, una ecocardiografía (ecografía cardiaca), una resonancia magnética cardiaca, un control diario del ECG y pruebas genéticas (cuando están indicadas).
8. ¿Cómo se trata el cGMP: es necesaria la cirugía o bastan las pastillas?
La medicación es la base del tratamiento. En casos graves, puede ser necesaria la cirugía o la ablación septal percutánea.
9. ¿Qué es un DCI y cuándo se coloca en cGMP?
Un DAI es un desfibrilador cardioversor implantable, un dispositivo que previene la muerte súbita por arritmias. Se coloca en pacientes de alto riesgo por indicación de un médico.
10. ¿Es posible curar completamente la GMPc? ¿Cuál es el pronóstico?
La PCM no puede curarse completamente, pero los síntomas pueden controlarse eficazmente. Con el enfoque adecuado, el pronóstico es favorable, sobre todo en ausencia de complicaciones graves.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.