Hirntumoren: Ätiologie, Klassifikation, Symptome, Diagnostik und Therapie
Gontsov A.Neuro-onkologischer Chirurg, MD
22 min lesen·Dezember 23, 2025
Dieser Artikel dient nur zu Informationszwecken
Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
Bei Hirntumoren (auch Gehirntumoren, Hirngeschwülste bzw. Gehirngeschwülste) handelt es sich um eine heterogene Gruppe von Neubildungen, die von verschiedenen Zellen des zentralen Nervensystems (ZNS) ausgehen oder als Folge von Metastasen aus anderen Organen (meist Lunge, Brustdrüse, Niere, bei Melanom) entstehen. Je nach Lokalisation und Wachstumsverhalten können Tumoren sowohl allgemeine zerebrale als auch fokale neurologische Symptome aufgrund von Kompression, Gewebsinfiltration und erhöhtem intrakraniellem Druck (ICP) verursachen.
Die meisten Hirntumoren treten sporadisch (d.h. zufällig) auf.
Selten sind sie genetisch (hereditäre bzw. erbliche Syndrome) bedingt (Neurofibromatose, Li-Fraumeni-Syndrom u. a.).
Als wichtigster nachgewiesener Risikofaktor gilt ionisierende Strahlung (hohe Strahlendosen).
Epidemiologie
Die Inzidenz von primären Hirn- und ZNS-Tumoren liegt bei etwa 25 Fällen pro 100.000 Einwohner pro Jahr. Werden alle Hirntumoren (primär und sekundär (Metastasen)) einbezogen, beträgt die Inzidenz über 45 Fälle pro 100.000 Einwohner (pro Jahr).
Etwa 30 % der primären (d. h. nicht metastatischen) Tumoren sind bösartig, der Rest ist gutartig.
Bei Patienten unter 40 Jahren ist die Wahrscheinlichkeit von primären, meist niedriggradig bösartigen Tumoren höher.
Zu den häufigsten primären Tumoren gehören Meningeome (zu mehr als 90 % gutartig) und Gliome (zu mehr als 80 % bösartig).
Nach dem 40. Lebensjahr treten metastatische Tumoren (mehr als 50 %) und Glioblastome (bösartigste Hirntumoren, die 50 % aller Gliome ausmachen) in den Vordergrund.
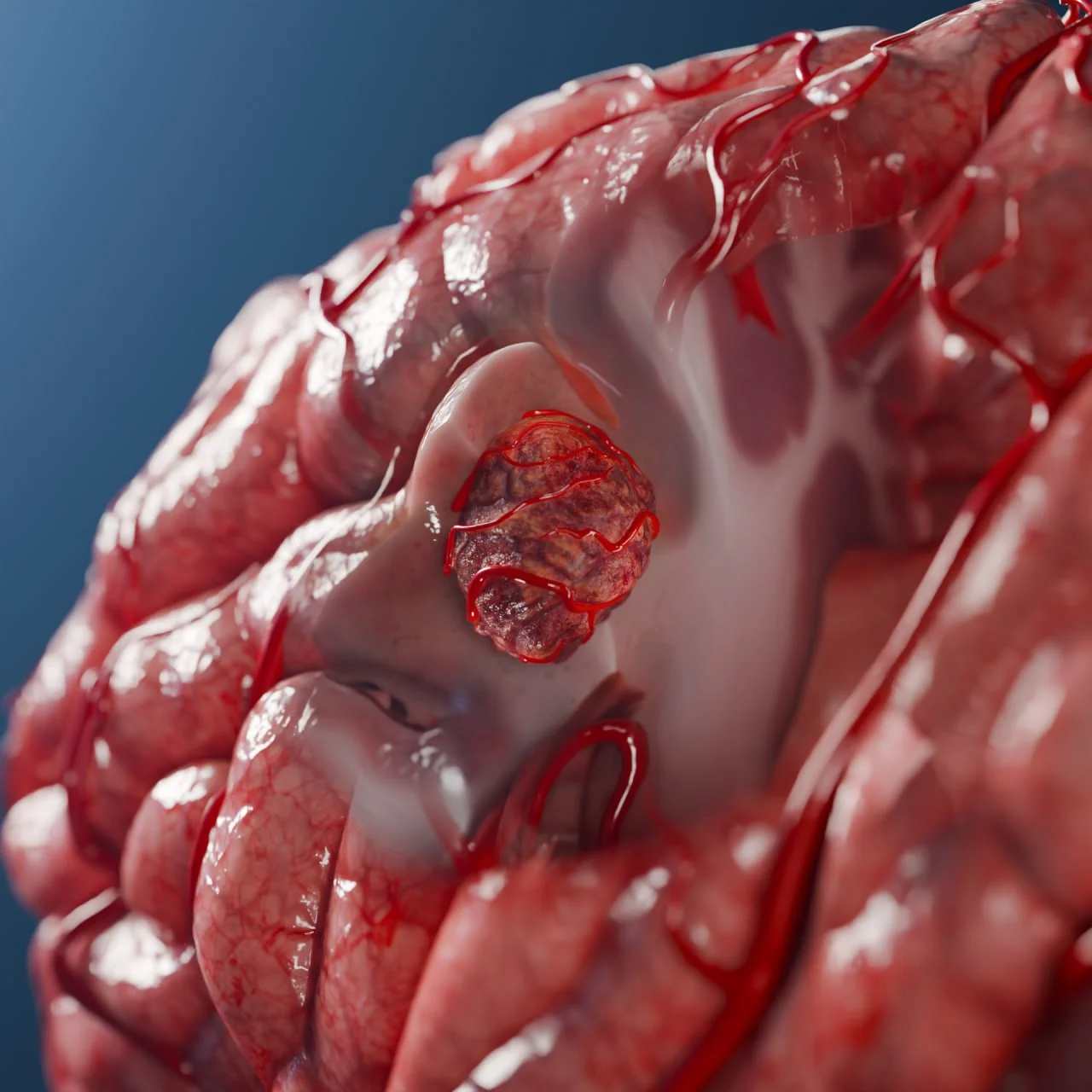
Hirnmetastase eines Nierenkarzinoms (linke Hemisphäre) – 3D-Modell
Bei Kindernsind ZNS-Tumoren die häufigste krebsbedingte Todesursache. Gliome (meist ein gutartiges pilozytisches Astrozytom mit einer 5-Jahres-Überlebensrate von > 90 %) >90%) sind die häufigste histologische Gruppe im Kindesalter, gefolgt von Medulloblastomen (die häufigsten bösartigen embryonalen ZNS-Tumoren bei Kindern mit einer 5-Jahres-Überlebensrate von 60–80 % mit Standardrisiko), Ependymomen, Glioblastomen, Kraniopharyngeomen, Keimzelltumoren usw.
Anatomie der Herpangina
In Bezug auf das Kleinhirnzelt
In Bezug auf das Kleinhirnzelt (Tentorium cerebelli) werden Tumoren in supratentorielle und infratentorielle unterteilt.
Supratentorielle Tumoren
Supratentorielle Tumoren(„oberhalb des Kleinhirnzeltes gelegen“) befinden sich oberhalb des Tentoriums in den Großhirnhemisphären, den Basalganglien, der Sehnervenkreuzung (Chiasma opticum), den Seitenventrikeln und dem 3. Ventrikel. Sie machen ~70–80 %der Tumoren bei Erwachsenen aus.
Beispiele:
Glioblastom
Astrozytom
Oligodendrogliom
Hypophysenadenom
Meningeom mit frontaler oder parietaler Lokalisation u. a.
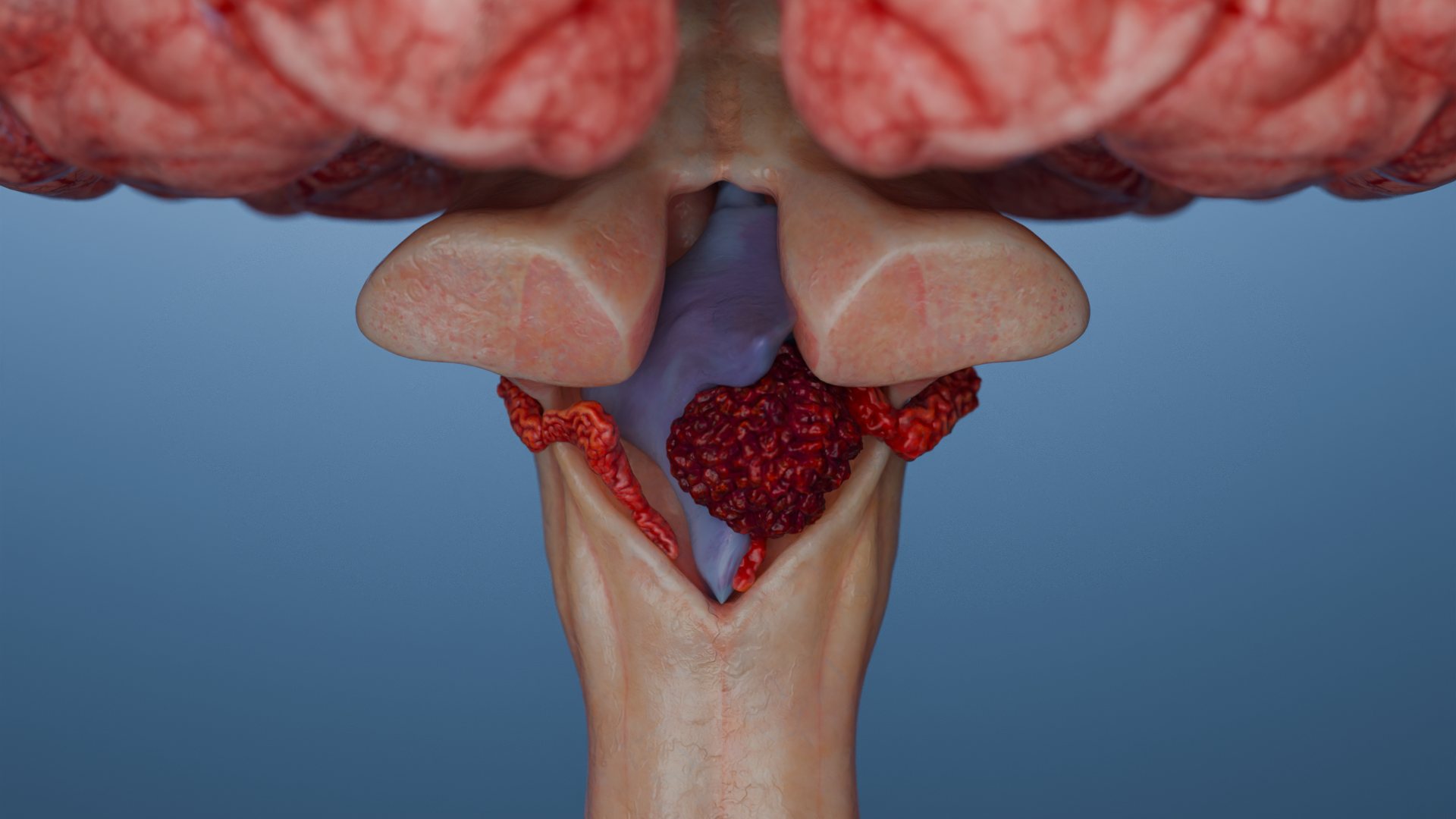
Infratentorielle Tumoren
Infratentorielle Tumoren („unterhalb des Kleinhirnzeltes gelegen“) befinden sich in der hinteren Schädelgrube (Fossa posterior cranii): Kleinhirn, Hirnstamm, 4. Ventrikel. Sie kommen vor allem bei Kindern vor (bis zu 60–70 % der Tumoren im Kindesalter).
In Bezug auf das Hirnparenchym („axiale“/zentrale ZNS-Struktur) können Tumoren in extraaxiale und intraaxiale (von lat. Axis) unterteilt werden.
Diese Begriffe spiegeln nicht nur die Lokalisation, sondern auch die Biologie des Wachstums, das klinische Verhalten und die Behandlungstaktik von Tumoren wider. Extraaxiale Tumoren sind meist gutartig, wobei intraaxiale in der Regel infiltrativ und bösartig sind.
Extraaxiale Tumoren
Extraaxiale Tumoren entstehen außerhalb des Hirnparenchyms, aber innerhalb des Schädels. Sie komprimieren oft das Gehirn, infiltrieren es aber nicht.
Beispiele:
Tumoren der Hirn- bzw. Rückenmarkshäute (Meningeom)
Tumoren der Hirnnerven (Schwannom)
Tumoren der Knochen der Schädelbasis (Chordom) u. a.
Intraaxiale Tumoren
Intraaxiale Tumoren entwickeln sich innerhalb der Hirnsubstanz aus dem Hirngewebe, häufiger aus Gliazellen (Astrozyten, Oligodendrozyten, Ependymzellen). Beispiele:
Glioblastom
Astrozytom
Ependymom u. a.
Klassifikation der Hirntumoren
Die aktuelle WHO-Klassifikation 2021 (WHO CNS 2021) legt den Schwerpunkt auf molekulargenetische Eigenschaften und weniger auf die reine Histologie.
Die Einteilung basiert auf drei Ebenen:
Histologie: Zellmorphologie und Tumorarchitektur.
Immunhistochemie: Proteinexpression (GFAP, p53, ATRX, Ki-67 u. a.)
Molekulare Marker: IDH1/2, 1p/19q, TP53, ATRX, H3, BRAF, CDKN2A/B, EGFR, TERT u. a.
Beispielsweise werden bei Gliomen zur Abklärung der Diagnose, Bestimmung der Prognose und Untersuchung des Ansprechens auf die Therapie folgende Faktoren untersucht:
MGMT (methyliert = gutes Ansprechen auf Temozolomid)
VEGF‑Expression(erhöht = sinnvoller Einsatz von Bevacizumab) u. a.
Der Übergang zur molekularen Klassifikation ermöglicht eine genauere Einteilung der Tumoren, eine präzisere Prognose des klinischen Krankheitsverlaufs und eine personalisierte Behandlung (Wahl der Chemotherapie, Vorhersage des Ansprechens auf die Therapie, Bestimmung der Angemessenheit einer Operation oder Bestrahlung).
Klinische Erscheinungen bei Hirntumoren
Allgemeine zerebrale Symptome:
Kopfschmerzen: bei 50–70 % der Patienten. Oft beidseitig, dumpf, bei Husten, Neigen und in der Nacht verstärkt
Krampfanfälle: 50–80 % bei primären Tumoren, insbesondere bei langsam wachsenden Tumoren (niedriggradige Gliome) und Metastasen, oft fokal
Erhöhter intrakranieller Druck: Trias (Kopfschmerzen, Übelkeit, Schwellung der Sehnervenpapille (auch Stauungspapille bzw. Papillenödem genannt)
Fokale neurologische Symptome:
Schwäche im Gesicht/in den Extremitäten: häufiger bei Läsionen des motorischen Kortex, möglicherweise reversibel durch Glukokortikoide
Sensibilitätsstörungen: sensomotorische Defizite sind nicht mit Dermatomen identisch
Aphasie (Schwierigkeiten bei der Wiedergabe oder dem Verstehen von Sprache): bei Läsionen der linken Hemisphäre, insbesondere im Broca- und Wernicke-Areal
Sehstörungen: Hemianopsie bei Betroffenheit der Sehbahn, Diplopie, verminderte Sehschärfe
Persönlichkeitsveränderungen: bei Lokalisation im Stirnlappen
Bei Kindern hängt die Symptomatik vom Alter, der Lage und der Art des Tumors ab. Die Manifestationen sind oft unspezifisch, was eine frühe Diagnose erschwert.
Indikationen für Neuroimaging (MRT/CT) bei Kindern mit Verdacht auf einen ZNS-Tumor:
anhaltende Kopfschmerzen > 4 Wochen, insbesondere mit morgendlichem Erbrechen, Bewusstseinsstörungen, bei Kindern < 4 Jahren
neue oder zunehmende neurologische Symptome (Krampfanfälle, Schwäche, Koordinations- und Sehstörungen)
Schielen, verminderte Sehschärfe, Stauungspapille
Regression der motorischen Fähigkeiten, Gangunsicherheit
Makrozephalie (Vergrößerung des Kopfes), insbesondere in Verbindung mit anderen Symptomen
3D-Animation — Plexus-chorioideus-Papillom
Diagnostik der Hirntumoren
Die Diagnose von Hirntumoren basiert auf der umfassenden Anwendung moderner Neuroimaging-Verfahren, histologischer und molekularer Verifizierung sowie zusätzlichen Untersuchungen bei unklarer Ätiologie oder Verdacht auf einen atypischen Verlauf.
Neuroimaging
MRT mit Kontrastmittel: Mittel der Wahl zur Erkennung und Klassifizierung von Hirntumoren. Damit lassen sich Größe, Lage, intrakranielle Ausbreitung, Vaskularisierung, Vorliegen von Nekrosen und Infiltrationsgrenzen beurteilen
Schädel-CT: bei Notfallindikationen oder Kontraindikationen für MRT
Schlecht differenzierte Gliome: Hyperintensität im T2-/FLAIR-Bild, kein Kontrastmittel-Enhancement, infiltratives Wachstum
Metastasen: rundlich, gut abgegrenzt, oft multipel, mit perifokalem Ödem
Meningeom: mit Hirnhäuten verbunden (Schädelbasis/Schädelkalotte/Großhirnsichel/Kleinhirnzelt), homogenes Kontrastmittel-Enhancement
Primäres ZNS-Lymphom: homogenes Kontrastmittel-Enhancement, deutliche Diffusionseinschränkungen (DWI), typisch für die periventrikuläre Region
Biopsie
Sie wird stereotaktisch oder chirurgisch durchgeführt, um die Diagnose und das molekulare Profiling (IDH, 1p/19q, MGMT, TP53, BRAF, H3K27 u. a.) zu bestätigen.
Glukokortikoide sollten (sofern zulässig) bei Verdacht auf ein Lymphom vorzugsweise vor der Biopsie abgesetzt werden, da der Tumor bei ihrem Einsatz vorübergehend schrumpft (Tumorregression).
Zusätzliche Diagnoseverfahren
PET-CT, MR-Spektroskopie, Perfusions-MRT: bei unklarer Ätiologie (Unterscheidung zwischen Tumor und Abszess, Demyelinisierung u. a.)
Lumbalpunktion:: bei Schädigung der Hirnhäute, Verdacht auf Meningitis, Enzephalitis, progressive multifokale Leukoenzephalopathie oder Lymphom. Bei ausgeprägter intrakranieller Hypertension ist keine Punktion angezeigt.
Differentialdiagnose
Zweck:
Unterscheidung zwischen Tumoren und nicht neoplastischen Prozessen (Infektion, Demyelinisierung, Gefäßerkrankungen)
Feststellung der primären oder metastatischen Natur des Tumors
Klärung des Histotyps, des molekularen Subtyps und der Bösartigkeit des Tumors
Ausschluss von Strahlennekrose und Abszessen nach der Behandlung
Aneurysma
Plötzliche Kopfschmerzen, im CT/MRT mit Kontrastmittel – vaskuläre Masse, in der Angiographie – erweiterte Arterie, Subarachnoidalblutung.
Schlaganfall
Plötzliches Auftreten, fokales neurologisches Defizit, ischämischer Bereich auf DWI/ADC-MRT sichtbar (Diffusionsrestriktion), häufiger über Gefäßbecken lokalisiert.
Enzephalitis
Subakutes Auftreten, Fieber, Verwirrtheit und Krampfanfälle (keine fokale Masse). Im MRT sind die Veränderungen in der Regel diffus, häufig in den Temporallappen (z. B. bei herpetischer Enzephalitis), ohne Masseneffekt.
Primärer Hirntumor
Allmählicher Beginn, fokale neurologische Symptome, Krampfanfälle, Kopfschmerzen. Im MRT-Bild sind üblicherweise infiltrative oder solide Masse, Schwellung, Masseneffekt und heterogene Kontrastmittelanreicherung erkennbar.
Metastasen
Oft multipel, kortikosubkortikal (Grenze zwischen grauer und weißer Substanz), mit ausgeprägtem Ödem und deutlichem ringförmigem Kontrastmittel-Enhancement, Tumorerkrankungen in der Anamnese.
ZNS-Lymphom
Im MRT-Bild ist homogenes Kontrastmittel-Enhancement und Lokalisation in tiefen Hirnstrukturen zu sehen, insbesondere bei Immunsuppression. Hohe Diffusionsbeschränkung (DWI+), mögliche schnelle Rückbildung nach Steroiden, daher ist es wichtig, diese vor der Biopsie zu vermeiden.
Hirnabszess
Häufig Fieber, Leukozytose und ein Infektionsherd (Mittelohrentzündung, Sinusitis, Bakteriämie). Im MRT-Bild ähnelt er einem Tumor, weist aber eine deutliche Diffusionseinschränkung auf (DWI+). Die MR-Spektroskopie zeigt spezifische Aminosäuren/Metaboliten.
Strahlungsnekrose
Sie tritt innerhalb von Monaten bis Jahren nach der Strahlentherapie auf und kann ein Tumorrezidiv vortäuschen. Unterscheidet sich durch MR-Perfusion und PET (Nekrose bedeutet Hypoperfusion, Hypometabolismus) und kann durch Steroide verringert werden.
Multiple Sklerose
Junges Alter, multifokale Läsionen in der weißen Substanz, FLAIR-hyperintens ohne Masseneffekt oder Kontrastmittelanreicherung; der klinische Verlauf ist rezidivierend mit Remissionen, häufig bei jungen Menschen.
Sinus-cavernosus-Thrombose
Akuter Beginn mit Kopfschmerzen (oft einseitig), Schwellung der Augenlider, Ophthalmoplegie und Sehstörungen. Die MRT-/MR-Venographie zeigt einen Thrombus in den venösen Sinus. Häufig kommt es zu Infektionen im Gesichts-, Nasen- und Sinusbereich.
Cluster-Kopfschmerz
Scharfe Schmerzen um das Auge herum, Tränenfluss, Nasenausfluss, die mehrere Minuten andauern, wiederholt auftreten und nicht mit neurologischen Ausfällen einhergehen. Keine strukturellen Veränderungen im MRT.
Idiopathischer Hydrozephalus
Häufig tritt bei älteren Menschen eine Trias von Symptomen (Gangstörungen, Demenz, Harninkontinenz) auf, möglicherweise begleitet von Kopfschmerzen und Übelkeit, im MRT zeigt sich eine Ventrikelerweiterung ohne fokale Massen.
Stoffwechselerkrankungen
Verursacht eher diffuse als fokale Symptome (Verwirrtheit, Krampfanfälle, Zittern), ohne fokale Masse im MRT. Allgemeine Tests (Glukose, Natrium, Leberenzyme, Kreatinin, Harnstoff usw.) helfen dabei, die Ursache zu identifizieren.
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Behandlung der Hirntumoren
Glukokortikoide
Sie werden bei Symptomen eines Hirnödems und eines erhöhten intrakraniellen Drucks eingesetzt (Dexamethason 8–16 mg täglich → 4 mg alle 6 Stunden).
Langfristige Anwendung sollte vermieden werden: Risiko von Komplikationen (Infektionen, Diabetes mellitus, Osteoporose) und geringerer Überlebensrate bei Patienten mit Glioblastomen.
Bei Verdacht auf ein Lymphom sind sie vor einer Biopsie kontraindiziert (wegen der durch Dexamethason verursachten Tumorschrumpfung).
Antiepileptika
Sie werden nur bei Vorliegen von Krampfanfällen verschrieben. Als Anfangstherapie wird Levetiracetam eingesetzt.
Die Prophylaxe vor chirurgischen Eingriffen erfolgt indikationsbezogen.
Chirurgische Behandlung
Eine Biopsie, Resektion (teilweise Entfernung) oder vollständige Entfernung ist bei gut zugänglichen Tumoren, die Symptome verursachen, bei unklarer Diagnose und akuter Dekompensation angezeigt.
Moderne Technologien wie präoperative MRT-Traktographie, Neuronavigation, intraoperative MRT und Ultraschall des Gehirns, Mikroskop, Fluoreszenzfarbstoff erhöhen die Radikalität und Sicherheit der Operation erheblich.
Strahlentherapie und medikamentöse Tumortherapie
Strahlentherapie (Bestrahlung) verdoppelt die postoperative Lebenserwartung von Patienten mit malignen Gliomen.
Stereotaktische Radiotherapie/Radiochirurgie (SRS) führt bei schwer zugänglichen/strahlenresistenten Tumoren häufig zu einer anhaltenden lokalen Kontrolle (das Tumorwachstum wird gestoppt).
Die Strahlentherapie und medikamentöse Tumortherapie hängen vom Tumortyp ab:
ZNS-Lymphome: Biopsie + Hochdosis-Chemotherapie mit Methotrexat ± Strahlentherapie.
Metastasen: SRS bzw. offene Chirurgie (Kraniotomie) bei einzelnen Herden (evtl. kombiniert mit neoadjuvanter SRS) + gelegentlich zielgerichtete Tumortherapie und Immuntherapie. Multiple Metastasen oder inoperable Prozesse: SRS bzw. Ganzkörperbestrahlung.
Meningeome: Überwachung (bei kleinen asymptomatischen Meningeomen), Operation und/oder Strahlentherapie (SRS).
Schwannome und Neurofibrome: Überwachung, Operation oder SRS.
Jedes Jahr gibt es mehr und mehr Möglichkeiten für die medikamentöse Behandlung von (gut- und bösartigen) Tumoren, aber die wichtigsten Therapieoptionen sind immer noch die chirurgische Resektion und die Strahlentherapie.
Besonderheiten der Behandlung von ZNS-Tumoren bei Kindern
Die chirurgische Entfernung ist die wichtigste und erste Behandlungsmethode (außer bei diffusen Hirnstammgliomen und einigen Tumoren im Bereich der Sehbahn). Häufig wird eine ventrikulo-peritoneale Shunt-Operation (kurz VP-Shunt-OP genannt) zur Kontrolle der intrakraniellen Hypertonie (bei Anzeichen von Hydrozephalus) durchgeführt.
Je nach Histologie und Alter kommt eine Strahlentherapie zum Einsatz (bei Kindern unter 3 Jahren wird diese wegen des Risikos neurokognitiver Störungen in der Regel vermieden).
Die Chemotherapie wird in Kombination mit chirurgischen Eingriffen und Strahlentherapie eingesetzt, insbesondere bei Kindern ab 3 Jahren mit embryonalen Tumoren (Medulloblastom), Ependymomen und einigen Formen von Gliomen. Bei Säuglingen und Kleinkindern wird sie eingesetzt, um die Strahlentherapie hinauszuschieben oder zu ersetzen, wodurch deren Toxizität verringert wird.
Die modernen Behandlungsansätze bei ZNS-Tumoren beruhen auf multidisziplinärer Zusammenarbeit, molekularer Stratifizierung und personalisierter Therapie.
Die Entwicklung von Strahlen- und Arzneimitteltherapie (einschließlich zielgerichteter Therapie und immunonkologischer Methoden) schreitet zügig voran, was auf bessere Behandlungsergebnisse in der Zukunft hoffen lässt.
Wichtig sind eine rechtzeitige Diagnostik, die richtige Wahl des chirurgischen Eingriffs und der postoperativen Therapie sowie eine umfassende Rehabilitation der Patienten.
FAQ
1. Was verursacht Hirntumoren und sind sie vererbbar?
Die meisten Hirntumoren treten sporadisch, d. h. zufällig auf. Selten sind sie genetisch durch hereditäre Syndrome wie Neurofibromatose bedingt. Der wesentliche nachgewiesene Risikofaktor ist die Einwirkung ionisierender Strahlung.
2. Wie äußern sich Hirntumoren und was sind ihre ersten Symptome bei Männern und Frauen?
Die Symptome eines Hirntumors unterscheiden sich nicht nach Geschlecht und können Kopfschmerzen, die morgens oder beim Husten stärker werden, Krampfanfälle und kognitive Störungen umfassen. Je nach Lokalisation sind fokale neurologische Symptome wie Schwäche in den Extremitäten, Aphasie oder Sehstörungen möglich.
3. Welche Anzeichen und Symptome eines Hirntumors treten bei Kindern typischerweise auf?
Die Symptomatik ist bei Kindern oft unspezifisch, was eine frühzeitige Diagnose erschwert. Zu den alarmierenden Symptomen zählen anhaltende Kopfschmerzen mit morgendlichem Erbrechen, Regression der motorischen Fähigkeiten, Gangstörung, Schielen sowie eine überdurchschnittliche Kopfgröße (Makrozephalie) bei Kleinkindern.
4. Wie sieht ein Hirntumor im MRT aus?
Eine KM-verstärkte MRT ist das wichtigste Diagnoseverfahren, wobei der Tumortyp dessen Erscheinungsbild maßgeblich beeinflusst. So weist beispielsweise ein Glioblastom häufig unregelmäßige Konturen und eine heterogene Kontrastmittelaufnahme mit zentraler Nekrose auf, während Metastasen in der Regel als rundliche, gut abgegrenzte Herde mit ausgeprägter Schwellung erscheinen.
5. Kann ein Hirntumor geheilt werden und wie wird er entfernt?
Dies hängt von der Form, Größe, Lokalisation und der molekularen Beschaffenheit des Tumors ab. Die Haupttherapie besteht in einer totalen bzw. partiellen chirurgischen Resektion. Moderne Technologien wie Neuronavigation und intraoperative MRT erhöhen die Radikalität und Sicherheit der Operation.
6. Wie schnell wächst ein Hirntumor und wie lange lebt man damit?
Die Wachstumsgeschwindigkeit und die Prognose hängen vom Malignitätsgrad des Tumors ab. Einige Tumoren, wie z. B. niedriggradige Gliome, können langsam wachsen, während Glioblastome sich rasch entwickeln. Die Lebenserwartung ohne oder bei unwirksamer Therapie ist äußerst gering, jedoch kann sie durch moderne komplexe Behandlungsmethoden wie Operationen, Strahlen- und medikamentöse Therapien deutlich erhöht werden.
7. Kann ein Hirntumor von selbst verschwinden und wie gefährlich ist er?
Ein Hirntumor heilt nicht spontan und erfordert medizinische Maßnahmen bzw. Verlaufskontrolle. Das Gefährliche ist, dass Tumoren eine Kompression und Infiltration des umliegenden Gewebes sowie einen Anstieg des Hirndrucks verursachen, wodurch allgemeine zerebrale und fokale neurologische Symptome auftreten. Unbehandelt kann das fatale Folgen haben.
Quellenverweise
1.
VOKA-Katalog. [Elektronische Quelle].
https://catalog.voka.io/
2.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021 Aug 2;23(8):1231-1251. doi: 10.1093/neuonc/noab106. PMID: 34185076; PMCID: PMC8328013.
3.
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol. 2023 Oct 4;25(12 Suppl 2):iv1-iv99. doi: 10.1093/neuonc/noad149. PMID: 37793125; PMCID: PMC10550277.
4.
Price M, Ballard C, Benedetti J, Kruchko C, Barnholtz-Sloan JS, Ostrom QT. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2018-2022. Neuro Oncol. 2025. doi: 10.1093/neuonc/noaf194.
5.
Smith HL, Wadhwani N, Horbinski C. Major Features of the 2021 WHO Classification of CNS Tumors. Neurotherapeutics. 2022 Oct;19(6):1691-1704. doi: 10.1007/s13311-022-01249-0. Epub 2022 May 16. PMID: 35578106; PMCID: PMC9723092.
6.
Bonneville F, Jäger HR, Smirniotopoulos JG. Differential Diagnosis of Intracranial Masses. 2024 Feb 11. In: Hodler J, Kubik-Huch RA, Roos JE, editors. Diseases of the Brain, Head and Neck, Spine 2024-2027: Diagnostic Imaging [Internet]. Cham (CH): Springer; 2024. Kapitel 8. Available from: https://www.ncbi.nlm.nih.gov/books/NBK608601/doi: 10.1007/978-3-031-50675-8_8.
7.
Grand S, Nedunchelian M, Charara S, Demaison R, Jean C, Galloux A, Kastler A, Attye A, Berthet C, Krainik A. Tumor or not a tumor: Pitfalls and differential diagnosis in neuro-oncology. Rev Neurol (Paris). 2023 Jun;179(5):378-393. doi: 10.1016/j.neurol.2023.03.011. Epub 2023 Apr 6. PMID: 37030987.
8.
Chinthala AS, Obeng-Gyasi B, Virgin KL, Deckert M, Mao G. Brain metastasis mimicking brain abscess: illustrative case and systematic review. J Neurosurg Case Lessons. 2025 Oct 20;10(16):CASE25528. doi: 10.3171/CASE25528. PMID: 41115317; PMCID: PMC12548541.
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io