Dilatative Kardiomyopathie: Ätiologie, Pathogenese, Symptome, Diagnose und Behandlungsmethoden
Kizyukevich O.Kardiovaskulärer Chirurg, MD
14 min lesen·November 13, 2025
Dieser Artikel dient nur zu Informationszwecken
Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
Die dilatative Kardiomyopathie (DCM) ist eine Myokarderkrankung, die durch eine Dilatation (Erweiterung) und systolische Dysfunktion des linken oder beider Ventrikel gekennzeichnet ist, ohne dass diese Veränderungen auf eine koronare Herzkrankheit, angeborene Missbildungen, Bluthochdruck oder Klappenanomalien zurückzuführen sind. Gemäß bevölkerungsbasierten Studien liegt die Prävalenz der DCM bei etwa 0,036–0,400 %.
3D-Animation: Dilatative Kardiomyopathie
Ätiologie
Die Ursachen der dilatativen Kardiomyopathie (DCM) sind äußerst heterogen und umfassen sowohl vererbte (genetische/familiäre) als auch erworbene Faktoren:
Genetische Ursachen
Die Erkrankung wird vorwiegend autosomal-dominant vererbt.
Bis zu 50 % der Fälle können familiär bedingt sein.
Die maßgeblichen Gene sind TTN, LMNA, FLNC, BAG3, DSP, RBM20, MYH7, SCN5A.
Kann in Verbindung mit Arrhythmien, Erregungsleitungsstörungen oder einem Überlappungsphänotyp (z. B. mit Anzeichen einer arrhythmogenen Kardiomyopathie, CM) auftreten.
Entzündliche (postmyokarditische) Ursachen
Häufig als Folge einer Virusinfektion (Parvovirus B19, HHV-6, Adenoviren, Enteroviren).
Auch autoimmune Faktoren kommen in Frage, assoziiert mit Krankheiten wie systemischem Lupus erythematodes, Sarkoidose, rheumatoider Arthritis usw.
Einfluss toxischer Substanzen
Alkohol weist eine unmittelbare Myokardtoxizität auf, insbesondere bei hohen Dosen und längerem Gebrauch.
Anhaltendes, unbehandeltes Vorhofflimmern, atriale oder ventrikuläre Tachykardie, paroxysmale Tachykardie.
Die Veränderungen sind durch Herzfrequenz- und Rhythmuskontrolle möglicherweise reversibel.
Peripartale Kardiomyopathie
Tritt in den letzten Monaten der Schwangerschaft oder innerhalb von 5 Monaten nach der Entbindung auf.
Fälle ohne bekannte Ursache
Es wird eine Ausschlussdiagnose gestellt, wenn keine sekundären Ursachen identifiziert werden und Gentests keine Informationen liefern.
Pathogenese der dilatativen Kardiomyopathie
Unabhängig von der Ursache sind die pathogenetischen Mechanismen ähnlich: Schädigung der Kardiomyozten, Entzündungsaktivierung, myokardiales Remodeling und eine fortschreitende Verschlechterung der kontraktilen Funktion.
Phasen der dilatativen Kardiomyopathie:
Primäre Schädigung der Kardiomyozten, die zur Aktivierung von Entzündungen führt:
Genetisch: Störungen in der Struktur von Sarkomer, Zellkern, Zytoskelett usw.;
Toxisch: Ansammlung freier Radikale, mitochondriale Dysfunktion, direkte Schädigung der Herzmuskelmembranen;
Viral: Chronische Entzündung und Fibrose als Folge einer autoimmunen Herzmuskelentzündung.
Störung des intrazellulären Kalziumstoffwechsels, des Energiestoffwechsels und Apoptose der Kardiomyozten.
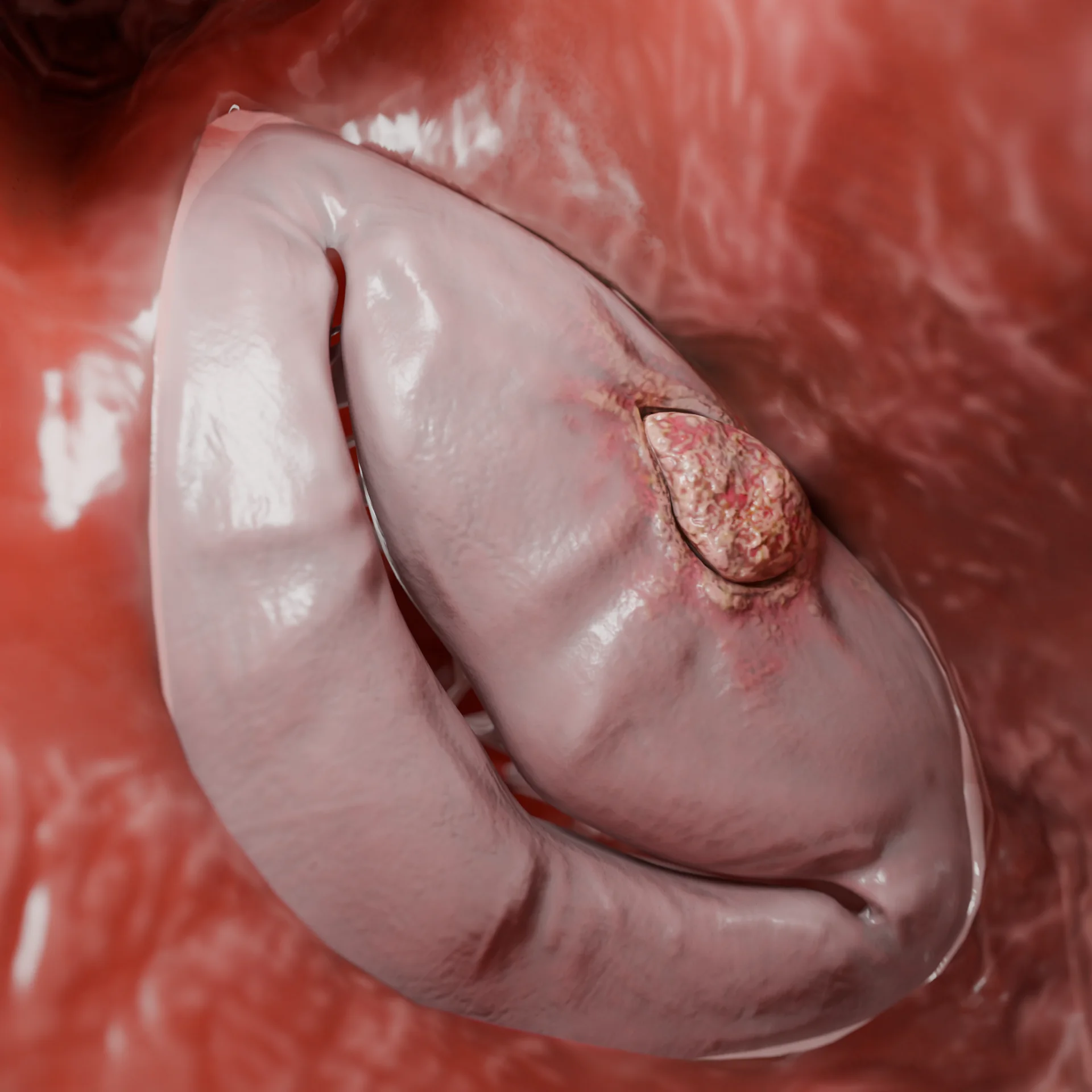
Umstrukturierung des Herzmuskels: Wandverdünnung, Dilatation der Herzhöhle (hauptsächlich des linken, seltener beider Ventrikel), Entwicklung einer interstitiellen Fibrose. Als Folge der Anulusdilatation und einer Dysfunktion des Papillarmuskels entwickelt sich häufig eine Atrioventrikularklappeninsuffizienz.
Die kurzfristig hämodynamisch günstige kompensatorische Aktivierung neurohormonaler Systeme (RAAS, sympathoadrenales System) kann langfristig zu einer dekompensierten Herzinsuffizienz führen.
Verdünnte linke Ventrikelwand und Erweiterung der Herzkammer: 3D-ModellMitralklappeninsuffizienz aufgrund von Dilatation der linken Herzkammer (LV) und des Klappenanulus: 3D-Modell
Symptome
Symptome der dilatativen Kardiomyopathie treten aufgrund einer fortschreitenden systolischen Dysfunktion und Blutstauung sowohl im systemischen als auch im Lungenkreislauf auf:
Belastungsdyspnoe, die schließlich zu Ruhedyspnoe fortschreitet;
Erschöpfung, reduzierte Belastbarkeit bei körperlicher Aktivität;
Orthopnoe und paroxysmale nächtliche Dyspnoe;
Schwellungen der unteren Extremitäten;
Vergrößerung der Leber, Aszites;
Ohnmacht, Schwindel (möglich bei Arrhythmien oder niedrigem Blutdruck);
Tachykardie, Rhythmusstörungen (insbesondere Vorhofflimmern und ventrikuläre Arrhythmien);
Seltener sind Schmerzen in der Brust aufgrund einer subendokardialen Ischämie.
Diagnose der dilatativen Kardiomyopathie
Die Diagnose einer DCM erfolgt durch den Nachweis einer linksventrikulären Dilatation und systolischen Dysfunktion, die nicht auf Ischämie, Bluthochdruck, Klappenfehlbildungen oder angeborene Pathologien zurückzuführen ist. Ziel der Diagnose ist es, den kardiomyopathischen Phänotyp zu bestätigen, die Ursachen zu bestimmen, das Ausmaß der Veränderungen der Herzstrukturen zu bewerten und die Wahrscheinlichkeit für die Entwicklung unerwünschter Ereignisse abzuschätzen.
Instrumentelle Methoden
Ein Herz-Ultraschall ist die Hauptmethode für die Primärdiagnose:
Das linksventrikuläre enddiastolische Volumen (LVEDV) ist erhöht (>150–180 ml oder indexiert >75 ml/m²);
Verminderte Ejektionsfraktion (<45 %);
Globale Hypokinesie ohne regionale Anomalien;
Häufig: eine Mitral- und Trikuspidalinsuffizienz, pulmonale Hypertonie und eine Dilatation des rechten Ventrikels (RV).
Herz-MRT mit Gadolinium (Late-Gadolinium-Enhancement):
LiefertInformationen zur Herzmuskelstruktur und Gewebemerkmale, wie Fibrosen, Ödeme oder Fettgewebseinlagerungen;
Typisches Muster bei DCM: Meso- oder subepikardiale Akkumulation von Gadolinium in der lateralen Wand oder im Septum;
Unerlässlich bei Verdacht auf Myokarditis, Sarkoidose und Speicherkrankheiten.
Wird durchgeführt, um eine koronare Herzkrankheit (KHK) bei Patienten über 35 Jahren oder bei vorhandenen Risikofaktoren auszuschließen;
Zwingend erforderlich bei typischen Brustschmerzen, regionalen Auffälligkeiten im Herz-Ultraschall oder Ansammlung von Gadolinium im Spätstadium.
Holter-Monitor oder Langzeit-EKG (24–72 Stunden):
Ventrikuläre Arrhythmien (VE, VT), Vorhofflimmern (AF), Tachykardie, Pausen, Blockierungen (besonders bei Verdacht auf LMNA-mutierte Form);
Hilft bei der Therapieauswahl und der Entscheidung für oder gegen ein ICD.
Myokardbiopsie (je nach Indikation):
Bei Verdacht auf: aktive Myokarditis, infiltrative Erkrankungen (Amyloidose, Sarkoidose);
Begrenzte Anwendung, erfordert genaue Messwerte und ein hohes Ausführungs-Level.
Labormethoden
BNP/NT-proBNP:
Dies ist der aussagekräftigste Biomarker für dekompensierte Herzinsuffizienz. Die Werte steigen proportional zum Grad der Volumen- und Drucküberlastung an. Hohe Werte deuten auf eine Dekompensation hin, während bei niedrigen Werten eine Herzinsuffizienz (HI) ausgeschlossen werden kann.
Kardialspezifische Troponine (I oder T):
Leichte Erhöhung ist bei aktiver Entzündung (z. B. Myokarditis) oder ausgeprägter myokardialer Dehnung möglich. Bei einem starken und akuten Anstieg muss ein Myokardinfarkt ausgeschlossen werden.
Schilddrüsenhormone (TTG, freies T3 und T4):
Eine Schilddrüsenunterfunktion kann zu einer systolischen Dysfunktion führen, während eine Schilddrüsenüberfunktion eine durch Tachykardie bedingte dilatative Kardiomyopathie verursachen kann.
Glukose und glykiertes Hämoglobin (HbA1c):
Diabetes mellitus steht im Zusammenhang mit der Entwicklung einer diabetischen Kardiomyopathie und verschlechtert zudem den Verlauf einer Herzinsuffizienz (HI).
Ermöglichen den Nachweis eines Eisenmangels oder einer Hämochromatose. Letzteres kann zu einer sekundären Kardiomyopathie mit fortschreitender linksventrikulärer Dysfunktion führen.
Entzündungs- und Autoimmunitätsmarker (antinukleäre Antikörper, Rheumafaktor, Antikardiolipin-Antikörper, etc.):
Durchgeführt bei Verdacht auf eine autoimmune oder systemisch entzündliche Natur der Erkrankung – wie systemischer Lupus erythematodes, Sklerodermie, Myokarditis, etc.
Angiotensin-konvertierendes Enzym, löslicher Interleukin-2-Rezeptor und Kalziumspiegel:
Kommen bei Verdacht auf kardiale Sarkoidose zum Einsatz. Besonders relevant bei Vorliegen von Leitungsstörungen oder unklaren infiltrativen Veränderungen im MRT.
Lebertests, Kreatinin, Elektrolyte:
Werden routinemäßig getestet, um systemische Manifestationen der Herzinsuffizienz festzustellen und die Behandlungsverträglichkeit zu beurteilen.
Gentests:
Indiziert bei familiärer Vorgeschichte von Kardiomyopathie, plötzlichem Herztod, Blocks, schwerer Dysfunktion in jungem Alter oder wenn keine sekundären Ursachen vorliegen. Es werden Panels für Gene verwendet, die mit DCM (dilatative Kardiomyopathie) assoziiert sind (am häufigsten TTN, LMNA, BAG3, FLNC, SCN5A, etc.).
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Behandlung der dilatativen Kardiomyopathie
Medikamentöse Behandlung
Die medikamentöse Therapieder DCM erfordert einen umfassenden und streng individuellen Ansatz, der die klinischen und funktionellen Merkmale des Patienten berücksichtigt.
Die wichtigsten Medikamentengruppen:
ACE-Hemmer/ARB/ARNI (Sacubitril/Valsartan): verbessern die Überlebensrate, reduzieren Krankenhausaufenthalte;
Betablocker (Bisoprolol, Carvedilol, Nebivolol): reduzieren die Mortalität;
Mineralokortikoid-Antagonisten: bei EF von <35 %;
SGLT2-Hemmer (wie Dapa-/Empagliflozin) verbessern die Prognose, unabhängig davon, ob ein Diabetes vorliegt;
Diuretika: bei Symptomen von Flüssigkeitsansammlung;
Ivabradin: bei einer Herzfrequenz >70 im Sinusrhythmus, falls Betablocker nicht ausreichen;
Antikoagulanzien: bei Vorhofflimmern, Vorhandensein von Blutgerinnseln, hoher Herzfrequenz.
Chirurgische Behandlung
Einsetzen medizinischer Geräte:
ICD (Kardioverter-Defibrillator): bei EF <35 %, NYHA-Klassen II-III und Risiko für ventrikuläre Tachykardien;
CRT-P/CRT-D (Resynchronisationstherapie): bei QRS >130 ms, EF <35 % und Sinusrhythmus.
Chirurgische Korrektur einer Mitralinsuffizienz (sekundär) im Falle von:
Funktioneller Mitralregurgitation Grad II bis III;
LVEF 30–50 %, LV DCD <70 mm (keine signifikante Dilatation des linken Ventrikels);
Symptome einer Herzinsuffizienz trotz medikamentöser Therapie;
Verfahren: Annuloplastie (Verkleinerung des Faserrings), Segelrekonstruktion, in manchen Fällen MitraClip (kathetergeführte Korrektur).
1. Kann eine dilatative Kardiomyopathie vollständig geheilt werden?
Eine vollständige Heilung ist nicht möglich, aber mit der richtigen Behandlung kann die Lebensqualität und die Lebenserwartung deutlich gesteigert werden.
2. Was ist der Unterschied zwischen idiopathischen und genetischen Formen der DCM?
Bei der idiopathischen Form ist keine Ursache erkennbar, während die genetische Form durch vererbte Genveränderungen ausgelöst wird.
3. Welche Ursachen kann eine DCM bei Menschen ohne vorherige Herzerkrankung haben?
Die Erkrankung kann durch unentdeckte genetische Ursachen, Infektionen, Giftstoffe, hormonelle Ungleichgewichte oder anhaltende Belastung ausgelöst werden.
4. Wie lebensbedrohlich ist DCM und worin liegen die Hauptkomplikationen?
Die dilatative Kardiomyopathie lebensbedrohlich und schreitet ohne Behandlung vor; eine rechtzeitige Therapie kann die Risiken jedoch deutlich senken. Die Gefährlichkeit der Krankheit basiert auf drei Hauptkomplikationen: einer fortschreitenden Herzschwäche mit Folge eines Multiorganversagens, lebensbedrohlichem Arrhythmien, die zum plötzlichen Herztod führen können, sowie Blutgerinnseln, die einen tödlichen Schlaganfall verursachen können.
Ja, bis zu 50 % der Fälle sind familiär bedingt. Für Angehörige werden ein EKG und eine Herzultraschalluntersuchung empfohlen.
7. Was versteht man unter einer „reduzierten Ejektionsfraktion“?
Dies ist ein Indikator für die Pumpfunktion des Herzens. Diese ist bei DCM aufgrund der Schwächung des Herzmuskels reduziert.
8. Wann sollte bei DCM ein Defibrillator (ICD) eingesetzt werden?
Bei schwer verminderter EF (<35 %) und einem Risiko für schwere Arrhythmien.
9. Dürfen Personen mit DCM Sport treiben?
Nur mäßiges Training in Absprache mit einem Kardiologen ist erlaubt.
10. Ist eine Schwangerschaft mit DCM möglich?
Ja, aber nur in stabilem Zustand und unter strenger medizinischer Überwachung – die Risiken hängen von der Schwere der Krankheit ab.
11. Was sind die Merkmale der dilatativen Kardiomyopathie bei Kindern?
Bei Kindern hängt die dilatative Kardiomyopathie häufiger mit einer früheren Herzmuskelentzündung oder bestimmten Erbkrankheiten zusammen. Das klinische Bild kann unspezifisch sein (Dyspnoe, Schwierigkeiten bei der Nahrungsaufnahme) und die Prognose ist häufig ernster als bei Erwachsenen.
Verweise
1.
VOKA-Katalog. [Elektronische Quelle].
https://catalog.voka.io/
2.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies (Richtlinien der Europäischen Gesellschaft für Kardiologie (ESC) zur Behandlung von Herzmuskelerkrankungen 2023). Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi: 10.1093/eurheartj/ehad194.
3.
Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches (Dilatative Kardiomyopathie: Ursachen, Mechanismen sowie aktuelle und zukünftige Therapieansätze). Lancet. 2023 Sep 16;402(10406):998-1011. doi: 10.1016/S0140-6736(23)01241-2.
4.
Gigli M, Stolfo D, Merlo M, et al. Pathophysiology of dilated cardiomyopathy: from mechanisms to precision medicine (Pathophysiologie der dilatativen Kardiomyopathie: von Mechanismen zur Präzisionsmedizin). Nat Rev Cardiol. 2025 Mar;22(3):183-198. doi: 10.1038/s41569-024-01074-2.
5.
Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature (Dilatative Kardiomyopathie: von epidemiologischen zu genetischen Phänotypen: Eine translationale Übersicht der aktuellen Literatur). J Intern Med. 2019 Oct;286(4):362-372. doi: 10.1111/joim.12944.
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io