Der Inhalt dieser Website, einschließlich Texten, Grafiken und anderen Materialien, dient ausschließlich Informationszwecken. Sie sind nicht als Rat oder Anleitung gedacht. Bitte konsultieren Sie Ihren medizinischen Betreuer, wenn es um Ihren speziellen Gesundheitszustand oder Ihre Behandlung geht.
Die hypertrophe Kardiomyopathie (HCM, auch familiäre hypertrophe Kardiomyopathie, FHC) ist eine primäre Erkrankung des Herzmuskels, die durch eine unerklärliche, meist asymmetrische Vergrößerung der linksventrikulären Wand ohne begleitende Dilatation der Herzkammer gekennzeichnet ist. Die Ursache der Erkrankung sind Genmutationen der am Aufbau des Sarkomers beteiligten Strukturproteine, was zu morphologischen, elektrischen und hämodynamischen Störungen führt.
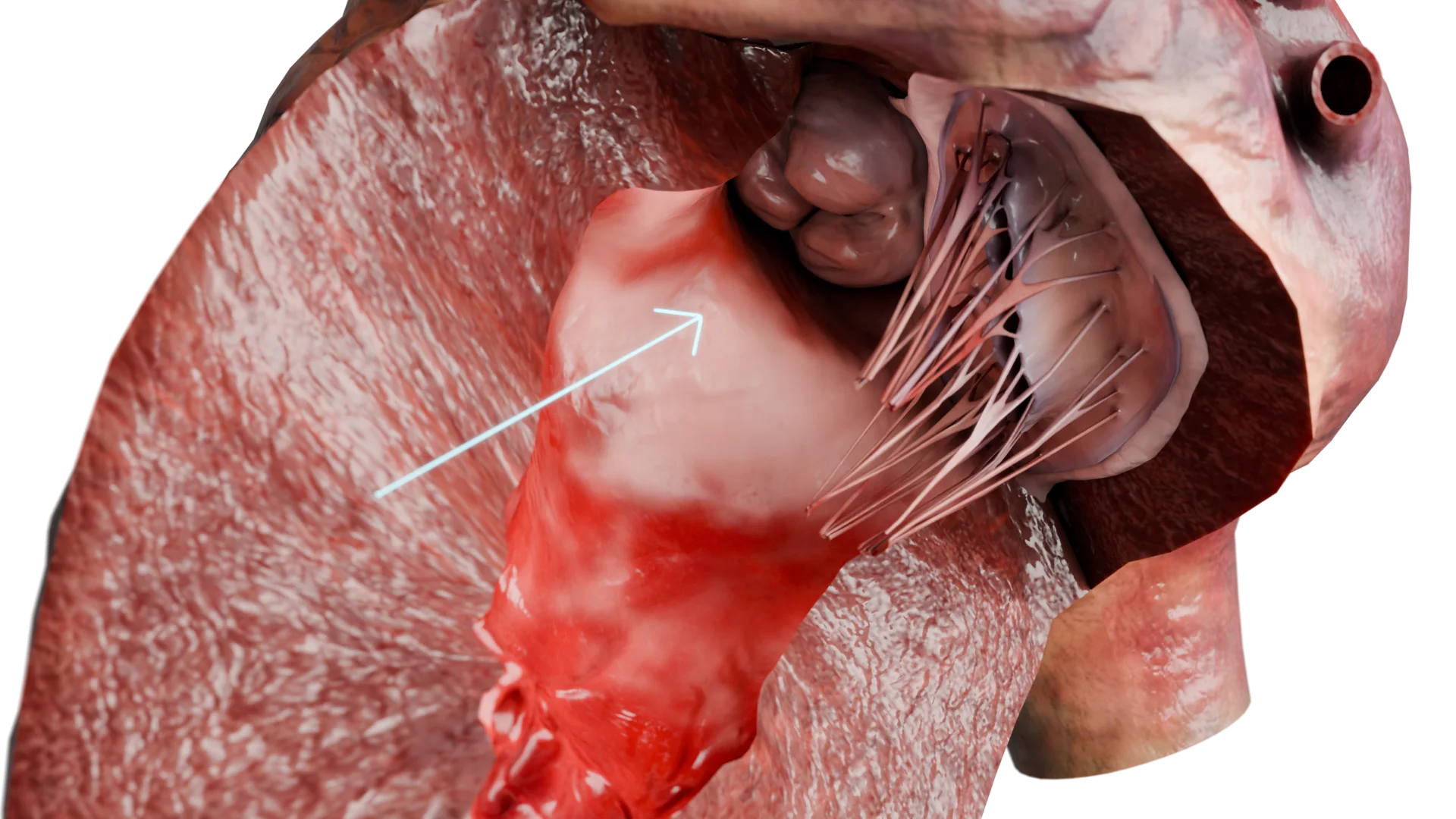
Verdickte Wand des linken Ventrikels bei HCM: 3D-Modell
HCM ist eine der häufigsten erblichen Formen der Kardiomyopathie und tritt bei etwa 1 von 500 Erwachsenen auf. Männer sind häufiger betroffen (das Verhältnis liegt bei etwa 3 : 2), jedoch wird die Erkrankung bei Frauen in der Regel erst später diagnostiziert und hat einen schwereren Verlauf.
Ätiologie
Die Krankheit wird durch eine gestörte Synthese und Funktion der kontraktilen Proteine des Sarkomers verursacht. In einigen Fällen werden aber auch sekundäre Formen festgestellt, die mit systemischen und metabolischen Störungen einhergehen.
Bis zu 60–70% der Fälle von hypertropher Kardiomyopathie stellen monogenetische Störungen dar.
Betroffene Gene
Gen
Protein
Häufigkeit von Mutationen
MYH7
β-Myosin schwere Kette
MYH7 und MYBPC3 machen 70% aller Mutationen aus
MYBPC3
Myosinbindendes Protein C
MYH7 und MYBPC3 machen 70% aller Mutationen aus
TNNT2
Troponin T (TnT)
Ca. 5%
TNNI3
Troponin I (TnI)
< 5%
TPM1
Tropomyosin
< 5%
Es handelt sich überwiegend um eine autosomal-dominante Erkrankung, d. h. ein verändertes Gen von einem Elternteil reicht aus. Dabei ist die Wahrscheinlichkeit der Erkrankung hoch, aber die Schwere und Form der Symptome können selbst innerhalb einer Familie stark variieren.
In etwa 30 % der Fälle tritt die Mutation zum ersten Mal auf, ohne familiäre Vorbelastung.
Einige Krankheitsbilder können die Klinik der hypertrophen Kardiomyopathie vortäuschen, pathogenetisch liegen ihnen aber ganz andere Ursachen zugrunde. Es ist äußerst wichtig, diese Fälle von der primären (sarkomeren) Form zu unterscheiden, da sie anders behandelt werden und eine andere Prognose haben.
Formen mit sekundärer Hypertrophie (HCM-Phänokopien)
Erkrankung
Mechanismus
Besonderheiten
Morbus Fabry (Fabry-Krankheit)
Genetisch bedingte, lysosomale Speicherkrankheit (ein Defekt des Enzyms alpha-Galaktosidase A)
Autosomal-rezessive Erbkrankheit, verursacht durch Mutationen im Gen FXN (mitochondriale Erkrankung)
Fortschreitende neurodegenerative Erkrankung (Ataxie, Muskelschwäche und -atrophie, Sprachstörungen u. a.)
Glykogenspeicherkrankheiten (Glykogenosen) (wie z. B. Morbus Pompe)
Glykogenablagerung in Zellenlysosomen, insbesondere in den Muskeln und im Herzen
Häufig ist die Skelettmuskulatur betroffen
Systemische arterielle Hypertonie
Reaktive Herzmuskelhypertrophie
I.d.R. symmetrisch, mit Hypertonus in der Vorgeschichte
Даже при наличии мутации в гене саркомерного белка клиническая выраженность и течение ГКМП зависят от дополнительных факторов:
epigenetische Regulatoren
begleitende arterielle Hypertonie
hohe körperliche Belastung (vor allem im Jugendalter)
Geschlecht und Hormonstatus (Frauen leiden häufiger unter obstruktiven Formen, die sich jedoch erst später manifestieren)
plötzlicher Herztod in der Familiengeschichte
Pathogenese
gestörte Sarkomerfunktion
Folgen der Genmutationen (s. Ätiologie):
Calcium-Überempfindlichkeit
verminderte Kontraktionsleistung
erhöhter Energiebedarf
Folge: Es entwickelt sich eine kompensatorische Herzmuskelhypertrophie. Vor allem ist das Ventrikelseptum im Bereich des linksventrikulären Ausflusstraktes (LVOT) betroffen.
Hypertrophie und gestörte Diastole (diastolische Dysfunktion)
Folgen der Wandverdickung:
verminderte LV-Compliance
beeinträchtigte LV-Füllung in der Diastole
erhöhter diastolischer Druck
Folge: Stauungslunge mit anschließendem Atemnot und anderen Symptomen einer Herzinsuffizienz mit erhaltener Ejektionsfraktion.
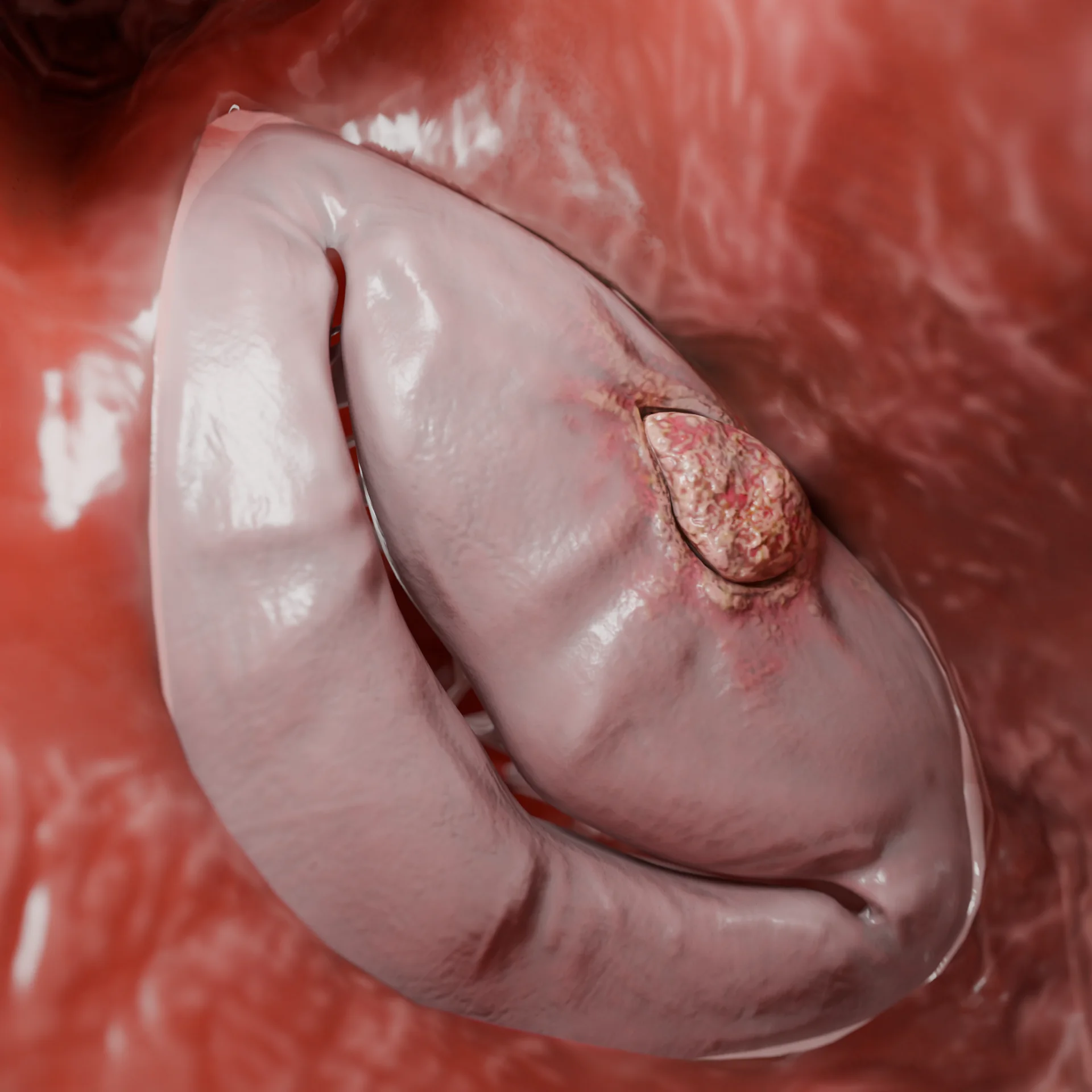
Ausgeprägte LVOT-Verengung (obstruktive Form)
Bei 60–70% der Patienten liegt eine LVOT-Obstruktion vor.
Durch den Venturi-Effekt wird das anteriore Mitralklappensegel zum Septum hin verschoben („Systolic Anterior Movement“, kurz SAM), der LVOT-Gradient erhöht sich und es kommt zu mitraler Regurgitation.
Folge: zunehmende hämodynamische Überlastung, die die Symptome verschlimmert und das Risiko von Arrhythmien erhöht.
LVOT-Obstruktion, bedingt durch eine Verdickung des Ventrikelseptums: 3D-Modell
Mikrovaskuläre Ischämie
Der hypertrophierte Herzmuskel benötigt mehr Sauerstoff, aber:
das Kapillarnetz kommt mit dem Wachstum des Gewebes nicht hinterher.
es besteht ein Ungleichgewicht zwischen Sauerstoffbedarf und Sauerstoffzufuhr.
häufig liegt eine Fibrose kleiner Gefäße vor.
Folge: Es kommt zu einer Myokardischämie bei ungestörter Koronardurchblutung mit anschließenden Brustschmerzen, Fibrose und einem erhöhten Risiko für Herzrhythmusstörungen.
Fibrose und elektrische Instabilität
Als Reaktion auf Ischämie und mechanische Myokardüberlastung bildet sich interstitielle und fokale Fibrose mit den Folgen wie:
Erregungsleitungsstörung
ventrikuläre Rhythmusstörungen
erhöhtes Risiko für plötzlichen Herztod (PHT)
3D-Animation: hypertrophe Kardiomyopathie
Symptome
Belastungsdyspnoe und schnelle Ermüdung
Thoraxschmerzen (Angina pectoris) ohne Schädigung der Koronararterien
Synkopen oder Präsynkopen (v.a. bei körperlicher Belastung)
ventrikuläre Rhythmusstörungen, Vorhofflimmern
PHT, insbesondere bei jungen Patienten und Leistungssportlern mit Obstruktion
Herzinsuffizienz: sowohl mit erhaltener systolischer linksventrikulärer Funktion (HFpEF), als auch mit reduzierter linksventrikulärer Ejektionsfraktion (HFrEF) in späten Stadien. Manifestiert sich durch Ödeme, Orthopnoe, Tachykardie, verminderte Belastbarkeit
Asymptomatischer Verlauf: Bei 25–30% der Patienten wird die Erkrankung bei einer Untersuchung aufgrund der Familienanamnese festgestellt. Schließt ein hohes Risiko für Komplikationen nicht aus.
Diagnostik der hypertrophen Kardiomyopathie
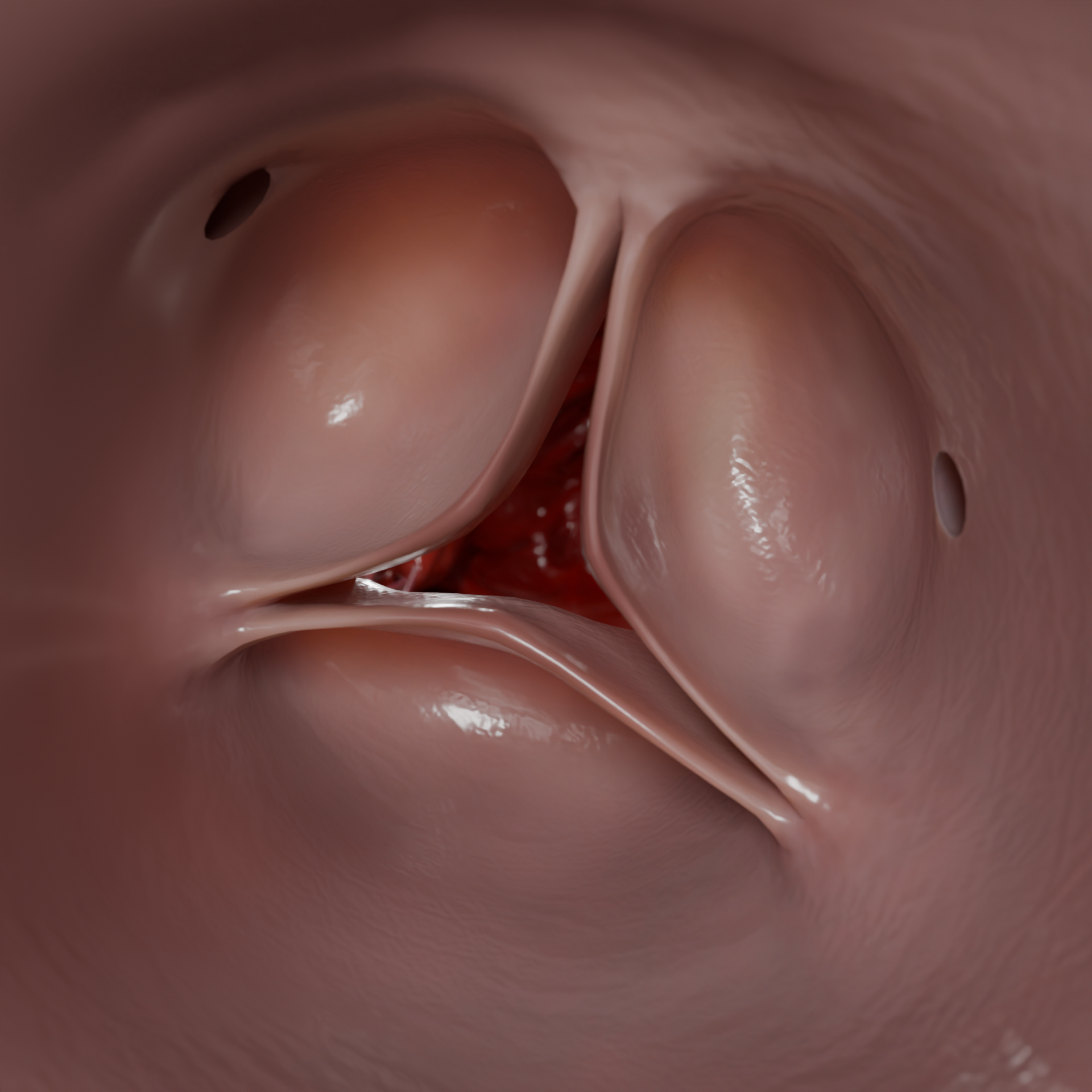
Transthorakale Echokardiographie (TTE) ist das Hauptverfahren für die Erstdiagnose. Befunde:
Myokarddicke. Die Diagnose ist wahrscheinlich bei einer Wanddicke von ≥ 15 mm bei Erwachsenen oder ≥ 13 mm bei Verwandten ersten Grades mit bestätigter HCM
Morphologie: asymmetrisch, konzentrisch, apikal
LVOT-Obstruktion (Gradient > 30 mmHg, ab 50 mmHg mit einer hämodynamischen Relevanz)
SAM (systolische Verschiebung des Mitralklappensegels) und mitrale Regurgitation
Funktion des linken Ventrikels und diastolische Dysfunktion
Abmessungen der Vorhöfe (insbesondere des linken Vorhofs wegen Vorhofflimmern)
YAKardiale Magnetresonanztomographie (Kardio-MRT) wird in folgenden Fällen empfohlen:
Mit der Echokardiographie lässt sich die Wanddicke nicht genau bestimmen.
Es besteht ein Verdacht auf apikale oder atypische HCM.
Eine Beurteilung der Myokardfibrose ist erforderlich.
Befunde:
Verteilung und Grad der Hypertrophie
Fibroseherde, die mit erhöhtem Risiko von Rhythmusstörungen und PHT assoziiert sind
Differenzierung mit Phänokopien (z. B. Amyloidose)
EKG (Elektrokardiogramm) ist unspezifisch, ist aber in über 90% der Fälle pathologisch. Linksventrikuläre Hypertrophiezeichen:
atypische Q-Zacke (in V4–V6, I, aVL) kann einen Myokardinfarkt vortäuschen
Einschätzung des Schweregrads von Rhythmusstörungen und Indikationen für eine ICD-Implantation
Belastungstest (Fahrradergometer oder Laufband). Indikationen:
Beurteilung der Belastbarkeit
Nachweis einer induzierten LVOT-Obstruktion
Gradientenbestimmung unter Belastung
Beurteilung ischämischer Symptomatik ohne Stenose der Koronararterien
Gentests. Empfohlen:
bei Patienten mit bestätigter HCM (insbesondere junge Patienten oder Patienten mit PHT in der Familienanamnese)
für das Screening der Verwanden ersten Grades (Kinder, Geschwister, Eltern)
bei Verdacht auf Phänokopien (Morbus Fabry, mitochondriale Erkrankungen u. a.)
Nachgewiesene Genmutationen: MYH7, MYBPC3, TNNT2, TNNI3, TPM1 kommen am häufigsten vor.
Labormarker:
NT-proBNP / BNP: erhöht bei Drucküberlastung und diastolischer Dysfunktion
Troponin T/I: kann bei mikrovaskulärer Ischämie mäßig erhöht sein
Weitere wissenschaftlich korrekte Inhalte finden Sie in unseren sozialen Medien
Abonnieren Sie und verpassen Sie nicht die neuesten Ressourcen
Behandlung der hypertrophen Kardiomyopathie
Anpassung des Lebensstils:
Vermeiden von schweren körperlichen Aktivitäten und Sport
Kontrolle des Blutdrucks und des Körpergewichts
Screening Verwandter ersten Grades
Medikamentöse Therapie:
β-Blocker (bevorzugt Bisoprolol, Metoprolol)
Verapamil – bei Kontraindikationen für β-Blocker
Disopyramid – als Ergänzungstherapie bei Obstruktion
Mavacamten — ein neuer Arzneistoff mit nachgewiesener Wirkung zur Senkung des Gradienten und zur Linderung der Symptome (gemäß ESC 2023 und AHA 2020)
Chirurgische Behandlung
Indikationen:
LVOT-Gradient ≥ 50 mmHg Hg. in Ruhe oder bei Belastung
schwere Symptomatik (NYHA III–IV), die auf β-Blocker, Verapamil oder Disopyramid nicht anspricht
ausgeprägte mitrale Regurgitation in Assoziation mit SAM
Erweiterte Septum-Myektomie:
Goldstandard der Chirurgie bei obstruktiver HCM
Der Eingriff erfolgt über Ministernotomie bzw. totale Sternotomie, in einigen Fällen auch über anterolaterale Minithorakotomie rechts.
Der hypertrophierte Abschnitt des Ventrikelseptums wird entfernt, wodurch der Druckgradient ausgeglichen wird.
Ggf. wird eine Mitralklappenplastik oder eine Resektion der sekundären Chordae durchgeführt.
Mögliche Komplikationen: Blockaden, Notwendigkeit einer ICD-Implantation, erneuter Gradientenaufstieg.
Transkoronare Ablation der Septumhypertrophie (TASH) (endovaskulärer Eingriff) kommt häufiger bei Patienten zum Einsatz, bei denen eine offene Operation kontraindiziert ist oder zu hohe Risiken birgt.
Ethanol wird in die perforierende Arterie injiziert→ künstlicher Infarkt → Verringerung der Wanddicke
Risiko eines kompletten AV-Blocks (bis zu 10%), der Patient sollte für die Implantation eines Herzschrittmachers bereit sein.
die Ergebnisse sind weniger vorhersagbar
Risiko einer inkompletten Beseitigung der Obstruktion
Implantation eines Kardioverter-Defibrillators (ICD)
Indikationen:
PHT in der Anamnese oder persistierende VT
LV-Wanddicke > 30 mm
PHT in der Familiengeschichte
Synkopen unklarer Ätiologie
LV-Ejektionsfraktion < 50% bei einem fortschreitenden Verlauf
Herztransplantation für Patienten mit Endstadium, einer refraktären Herzinsuffizienz, trotz Behandlung.
FAQ
1. Was ist eine hypertrophe Kardiomyopathie?
Die hypertrophe Kardiomyopathie (HCM) ist eine Erkrankung, bei der sich der Herzmuskel (in der Regel das Ventrikelseptum) abnormal verdickt. Anders als bei Hypertrophie aufgrund von Bluthochdruck oder Herzklappenfehlern ist die Verdickung bei HCM auf genetische Mutationen zurückzuführen und nicht auf eine Überlastung des Herzens.
2. Ist HCM vererbbar? Sollten Verwandte untersucht werden?
Ja, in den meisten Fällen wird HCM autosomal-dominant vererbt. Es wird empfohlen, Verwandte ersten Grades untersuchen zu lassen.
3. Welche Symptome können auf eine HCM hinweisen und wann sollte man einen Arzt aufsuchen?
Zu den Symptomen gehören Atemnot, Brustschmerzen, Schwindel und Synkopen, insbesondere bei körperlicher Belastung. Bei solchen Beschwerden oder der familiären Vorgeschichte eines plötzlichen Herztodes sollte man einen Kardiologen aufsuchen.
4. Ist HCM gefährlich? Kann man damit lange leben?
Bei rechtzeitiger Diagnose und angemessener Behandlung führen die meisten Patienten ein normales Leben. Allerdings erhöht die HCM in einigen Fällen das Risiko für Herzrhythmusstörungen und plötzlichen Tod, insbesondere wenn die Erkrankung unbehandelt bleibt.
5. Wodurch unterscheidet sich die obstruktive Form der HCM von der nicht-obstruktiven?
Bei der Obstruktion behindert die verdickte Scheidewand den Blutabfluss aus dem linken Ventrikel, was stärkere Symptomatik verursacht. Wenn keine Obstruktion vorliegt, ist der Abfluss nicht beeinträchtigt.
6. Darf man Sport treiben, wenn man HCM hat?
Intensive und Wettkampfsportarten sind bei HCM nicht zu empfehlen. In Absprache mit dem behandelnden Arzt sind moderate körperliche Aktivitäten zulässig.
7. Welche Untersuchungen sind für die Diagnose einer HCM erforderlich?
In der Regel werden ein EKG, eine Echokardiographie (Herzultraschall), eine Kardio-MRT, ein Langzeit-EKG und genetische Tests (sofern angezeigt) durchgeführt.
8. Wie wird HCM behandelt? Ist ein Eingriff erforderlich oder reichen Tabletten aus?
Die Behandlung erfolgt hauptsächlich mit Medikamenten. In schweren Fällen kann eine Operation oder eine transkutane Ablation der Septumhypertrophie erforderlich sein.
9. Was ist ein ICD und wann wird er bei HCM implantiert?
ICD steht für implantierbaren Kardioverter-Defibrillator. Dies ist ein Gerät, das den plötzlichen Tod durch eine Rhythmusstörung verhindert. Er wird Patienten mit hohem Risiko auf Empfehlung des Arztes implantiert.
10. Kann HCM vollständig geheilt werden? Wie ist die Prognose?
Die HCM kann nicht vollständig geheilt werden, aber die Symptomatik lässt sich wirksam unter Kontrolle bringen. Bei entsprechender Therapie ist die Prognose günstig, insbesondere wenn keine schweren Komplikationen auftreten.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.
St. Petersburg FL 33702, 7901 4th St N STE 300, USA
Ich danke Ihnen!
Ihre Nachricht wird gesendet! Unsere Experten werden sich in Kürze mit Ihnen in Verbindung setzen. Wenn Sie weitere Fragen haben, kontaktieren Sie uns bitte unter info@voka.io