Cardiomyopathie hypertrophique : étiologie, pathogénie, symptômes, diagnostic et méthodes de traitement
Kizyukevich O.Chirurgien cardiovasculaire, MD
14 minutes de lecture·juillet 22, 2025
Cet article est uniquement destiné à des fins d'information
Le contenu de ce site web, y compris les textes, les graphiques et autres matériels, est fourni à titre d'information uniquement. Il ne s'agit pas d'un avis ou d'un conseil. En ce qui concerne votre état de santé ou votre traitement spécifique, veuillez consulter votre fournisseur de soins de santé.
La cardiomyopathie hypertrophique (CMH) est une maladie primaire du myocarde caractérisée par une hypertrophie inexpliquée de la paroi du ventricule gauche, le plus souvent asymétrique, sans dilatation cavitaire. À la base de la maladie se trouvent des mutations des gènes codant les protéines sarcomériques, entraînant des altérations morphologiques, électriques et hémodynamiques.
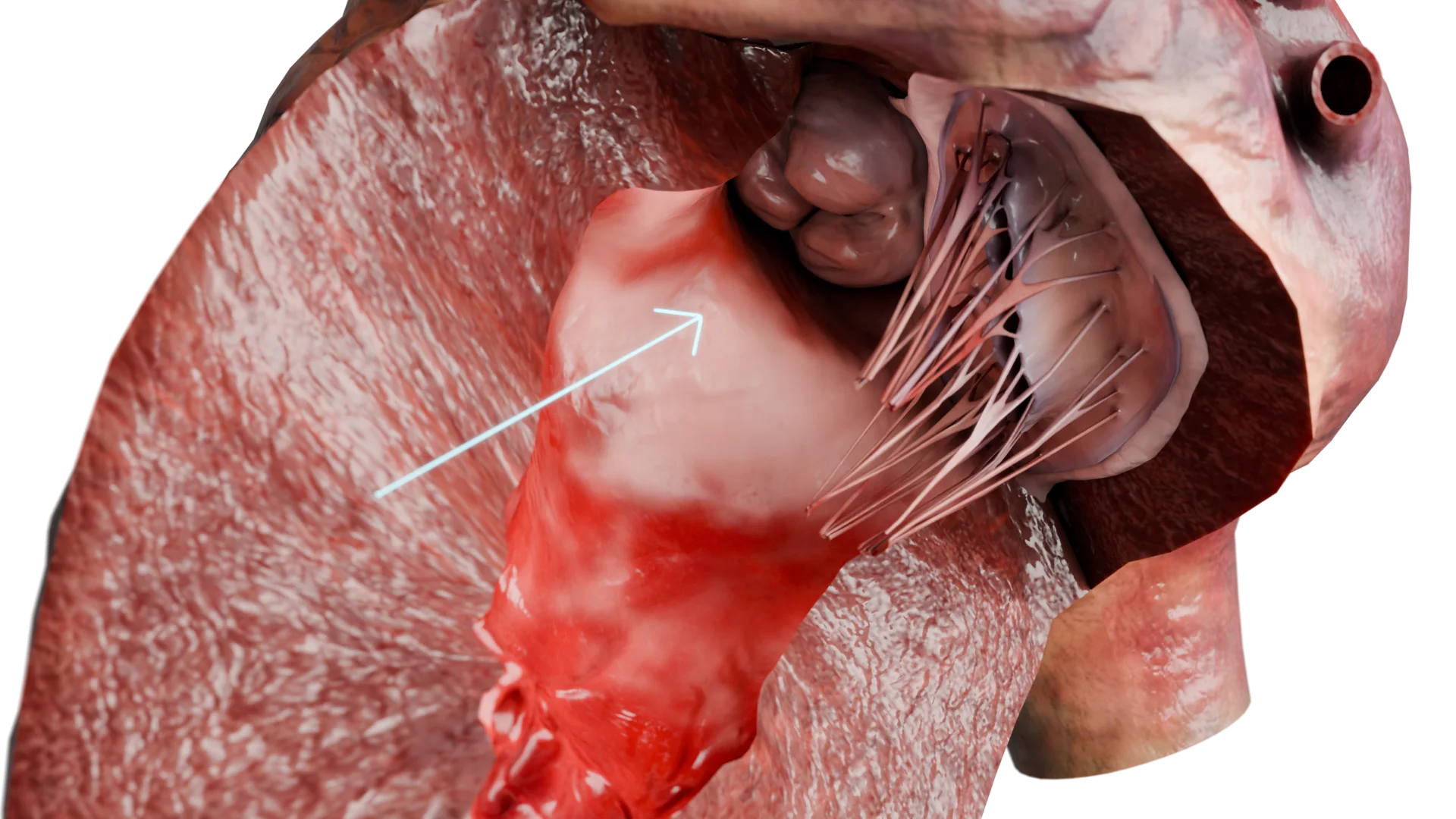
Paroi ventriculaire gauche épaissie dans la CMH – modèle 3D
La CMH est l’une des formes héréditaires les plus fréquentes de cardiomyopathies et touche environ 1 adulte sur 500. Les hommes sont plus souvent atteints que les femmes (ratio approximatif 3:2), mais chez les femmes la maladie est généralement diagnostiquée plus tardivement et évolue de façon plus sévère.
Étiologie
À la base de la maladie se trouve un trouble de la synthèse et de la fonction des protéines contractiles du sarcomère, mais dans certains cas sont identifiées des formes secondaires associées à des atteintes systémiques ou métaboliques.
Jusqu’à 60–70 % des cas de cardiomyopathie hypertrophique ont une étiologie monogénique.
Gènes associés au développement de la CMH
Gène
Protéine
Fréquence des mutations
MYH7
chaîne lourde β du myosine
MYH7 et MYBPC3 représentent environ 70 % des cas
MYBPC3
protéine C liant la myosine
MYH7 et MYBPC3 représentent environ 70 % des cas
TNNT2
Troponine T
Environ 5 %
TNNI3
Troponine I
<5%
TPM1
Tropomyosine
<5%
La maladie est transmise selon un mode autosomique dominant, c’est-à-dire qu’une seule copie mutée d’un parent suffit. La pénétrance est élevée, mais la gravité et les formes de manifestation peuvent varier fortement même au sein d’une même famille.
Dans environ 30 % des cas, la mutation apparaît de novo, sans anamnèse familiale.
Certaines maladies peuvent mimer le tableau clinique de la cardiomyopathie hypertrophique tout en ayant une origine pathogénique différente. Il est extrêmement important de les distinguer de la forme primaire (sarcomérique), car le traitement et le pronostic diffèrent.
Variantes avec hypertrophie secondaire (phénocopies de la CMH)
Maladie
Mécanisme
Particularités
Maladie de Fabry
Maladie lysosomale héréditaire de surcharge (déficit en α-galactosidase A)
Épaississement des parois, réduction de la contractilité, dysfonction diastolique
Ataxie de Friedreich
Maladie autosomique récessive par mutations du gène FXN (maladie mitochondriale)
Neurodégénérescence progressive (ataxie, faiblesse et atrophie musculaire, troubles de la parole, etc.)
Glycogénoses (ex. maladie de Pompe)
Accumulation de glycogène dans les lysosomes des cellules, en particulier dans les muscles squelettiques et le myocarde
Souvent atteinte de la musculature squelettique
Hypertension artérielle systémique
Hypertrophie myocardique réactionnelle
Généralement symétrique, avec antécédents d’hypertension
Даже при наличии мутации в гене саркомерного белка клиническая выраженность и течение ГКМП зависят от дополнительных факторов:
Régulateurs épigénétiques ;
Hypertension artérielle concomitante ;
Exercice physique de haute intensité (surtout à l’adolescence) ;
Sexe et statut hormonal (les femmes présentent plus souvent des formes obstructives mais avec manifestation plus tardive) ;
Antécédents familiaux de mort subite cardiaque.
Pathogenèse
Dysfonction sarcomérique
Les mutations génétiques (voir étiologie) entraînent :
Hypersensibilité au calcium ;
Diminution de l’efficacité contractile ;
Augmentation des besoins énergétiques.
Conséquence : hypertrophie myocardique compensatrice, prédominant au niveau de la cloison interventriculaire, en particulier dans la voie d’éjection du ventricule gauche (VEVG).
Hypertrophie et altération de la relaxation (dysfonction diastolique)
L’épaississement des parois entraîne :
Réduction de la compliance du ventricule gauche ;
Altération du remplissage diastolique ;
Élévation de la pression diastolique.
Conséquence : stase dans la petite circulation, se manifestant par dyspnée et autres symptômes d’insuffisance cardiaque à fraction d’éjection conservée.
Rétrécissement marqué de la voie d’éjection du VG (forme obstructive)
Chez 60–70 % des patients se forme une obstruction de la VEVG.
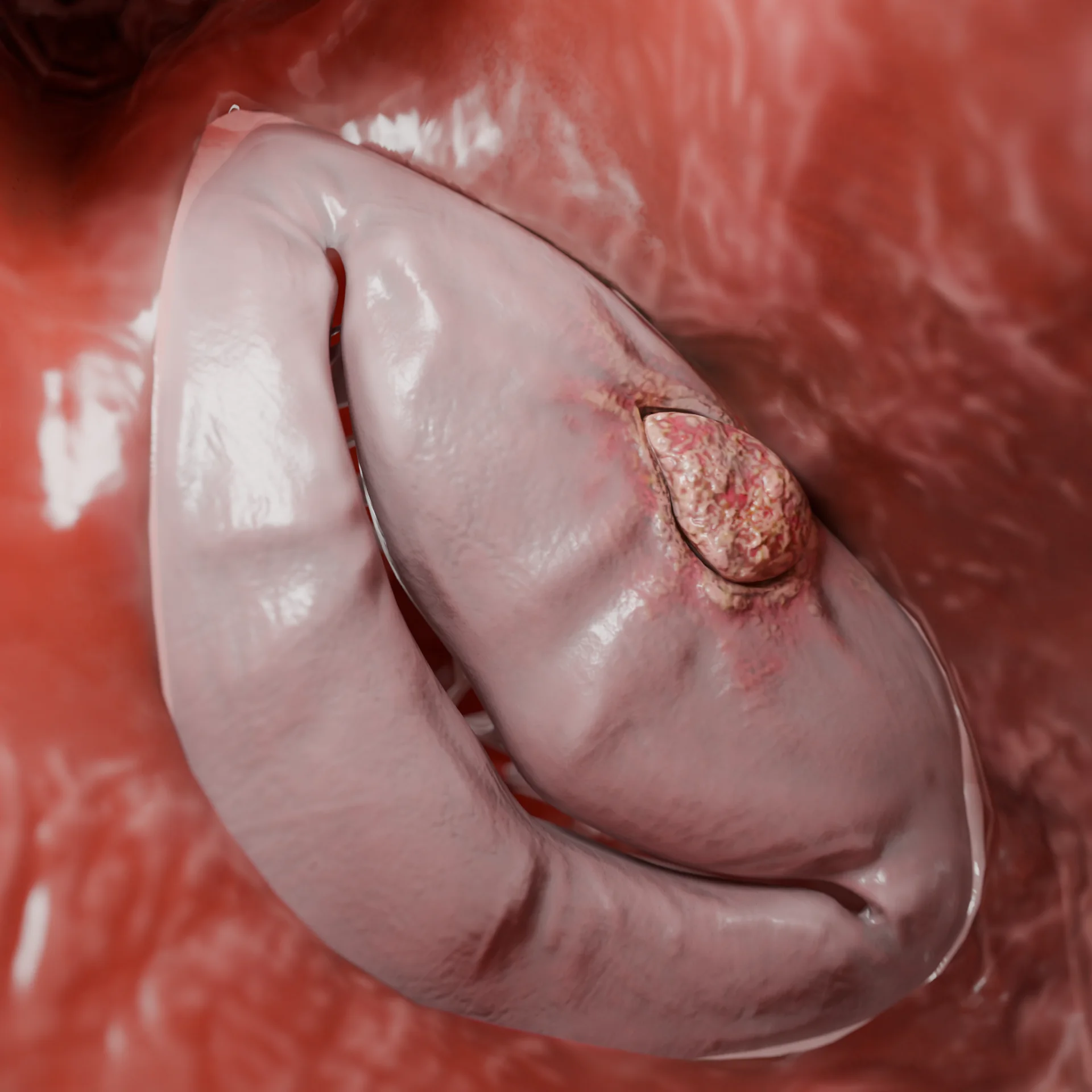
Par effet Venturi en systole : attraction de la grande valve mitrale vers la cloison (phénomène SAM), aggravation du gradient de pression, apparition d’une régurgitation mitrale.
Conséquence : surcharge hémodynamique croissante, aggravation des symptômes et augmentation du risque d’arythmies.
Obstruction de la VEVG par épaississement de la cloison interventriculaire – modèle 3D
Ischémie microvasculaire
Le myocarde hypertrophié nécessite plus d’oxygène, mais :
Le réseau capillaire ne suit pas la croissance tissulaire ;
Déséquilibre entre besoins et apport en oxygène ;
Fibrose fréquente des petits vaisseaux.
Conséquence : ischémie myocardique malgré artères coronaires normales, responsable de douleurs thoraciques, fibrose et risque accru d’arythmies.
Fibrose et instabilité électrique
En réponse à l’ischémie et à la surcharge mécanique se développe une fibrose interstitielle et focale, entraînant :
Troubles de la conduction ;
Arythmies ventriculaires ;
Risque accru de mort subite cardiaque (MSC).
Animation 3D – cardiomyopathie hypertrophique
Manifestations cliniques
Dyspnée d’effort, fatigue ;
Douleur thoracique (angor) sans lésion coronarienne ;
Syncopes ou états pré-syncopaux (surtout à l’effort) ;
Arythmies ventriculaires, fibrillation atrirale ;
MSC – particulièrement chez les jeunes patients et sportifs porteurs de forme obstructive ;
Insuffisance cardiaque : peut être à fraction d’éjection conservée (ICFEjC) ou réduite aux stades tardifs. Œdèmes, orthopnée, tachycardie, baisse de tolérance à l’effort ;
Forme asymptomatique : chez 25–30 % des patients, la maladie est découverte lors du dépistage familial réalisé en raison des antécédents familiaux. N’exclut pas un risque élevé de complications.
Diagnostic de la cardiomyopathie hypertrophique
Échocardiographie transthoracique (ETT)– méthode clé du diagnostic initial. Permet :
D’évaluer l’épaisseur myocardique. Diagnostic probable si épaisseur paroi ≥ 15 mm chez l’adulte ou ≥ 13 mm chez apparentés au 1er degré avec CMH confirmée ;
De déterminer la répartition de l’hypertrophie : asymétrique, concentrique, apicale ;
De détecter l’obstruction de la VEVG (gradient ≥ 30 mmHg, cliniquement significatif ≥ 50 mmHg) ;
Mettre en évidence le phénomène SAM (attraction systolique antérieure de la valve mitrale) et la régurgitation mitrale.
D’évaluer la fonction VG et la présence de dysfonction diastolique ;
De mesurer les dimensions des oreillettes (surtout gauche – risque de FA).
IRM cardiaque recommandée si :
L’ETT ne permet pas d’évaluer précisément l’épaisseur des parois ;
Suspicion de forme apicale ou atypique ;
Nécessité d’évaluer la fibrose myocardique.
Détecte :
Répartition et degré d’hypertrophie ;
Zones de fibrose – associées à un risque accru d’arythmies et de MSC ;
Différenciation avec les phénocopies (ex. amylose).
ECG – non spécifique mais anomalies présentes chez > 90 % des patients. Signes d’hypertrophie ventriculaire gauche :
Ondes Q atypiques (dérivations V4–V6, I, aVL) – pouvant mimer un infarctus ;
Modifications du segment ST et inversion de T ;
Troubles du rythme : FA, ESV, TV ;
Blocs auriculo-ventriculaires ou intraventriculaires.
Monitorage Holter. Indications :
Suspicion d’arythmies (ESV, TV, FA) ;
Syncopes ou lipothymies ;
Évaluation de la sévérité des troubles rythmiques et indications au DAI.
Test d’effort (tapis roulant ou vélo-ergométrie). Réalisé pour :
Évaluer la tolérance à l’effort ;
Détecter une obstruction induite ;
Mesurer le gradient sous effort ;
Identifier des symptômes ischémiques sans sténose coronarienne.
Tests génétiques . Recommandé :
Chez les patients avec CMH confirmée (surtout jeunes ou antécédents familiaux de MSC) ;
Dépistage des apparentés au 1er degré (enfants, frères/sœurs, parents) ;
Suspicion de phénocopie (maladie de Fabry, maladies mitochondriales, etc.).
Gènes les plus fréquemment testés : MYH7, MYBPC3, TNNT2, TNNI3, TPM1.
Marqueurs biologiques :
NT-proBNP / BNP – élevés en cas de surcharge de pression, dysfonction diastolique ;
Troponine T/I – peut être modérément élevée en cas d’ischémie microvasculaire.
Retrouvez d’autres contenus scientifiquement exacts sur nos médias sociaux
Abonnez-vous et ne manquez pas les dernières ressources
Traitement de la cardiomyopathie hypertrophique
Modification du mode de vie :
Exclusion des efforts physiques intenses et du sport de compétition ;
Contrôle de la pression artérielle et du poids corporel ;
Vérapamil – en cas de contre-indication aux β-bloquants ;
Disopyramide – en ajout chez les formes obstructives ;
Mavacamten – nouveau médicament avec effet démontré de réduction du gradient et amélioration des symptômes (ESC 2023 et AHA 2020).
Traitement chirurgical
Indications :
Gradient dans la voie d’éjection du ventricule gauche (VEVG) ≥ 50 mmHg. mmHg. au repos ou provoqué ;
Symptômes sévères (NYHA III–IV) réfractaires aux β-bloquants, vérapamil ou disopyramide ;
Régurgitation mitrale importante liée au phénomène SAM.
Myectomie septale élargie :
Traitement chirurgical de référence de la CMH obstructive ;
Réalisée par ministernotomie ou sternotomie complète, parfois par minithoracotomie antérieure droite ;
Résection d’un segment de cloison interventriculaire hypertrophiée éliminant le gradient ;
Plastie de la valve mitrale ou résection de cordages secondaires si nécessaire ;
Complications possibles : blocs de conduction, nécessité de DAI, récidive du gradient.
Ablation septale alcoolique (intervention endovasculaire) utilisée préférentiellement chez les patients pour qui la chirurgie à cœur ouvert est contre-indiquée ou à haut risque.
Injection d’éthanol dans une artère septale perforante → infarctus local → réduction de l’épaisseur septale ;
Risque de bloc AV complet (jusqu’à 10 %) – nécessité de préparation à l’implantation d’un stimulateur ;
Résultats moins prévisibles ;
Possibilité d’élimination incomplète de l’obstruction.
Implantation d’un défibrillateur automatique (DAI)
Indications :
Antécédent de MSC ou TV soutenue ;
Épaisseur paroi VG >30 mm ;
Histoire familiale de MSC ;
Syncopes d’étiologie indéterminée ;
FEVG <50% en cas d’évolution progressive.
Transplantation cardiaque chez les patients au stade terminal avec insuffisance cardiaque réfractaire malgré le traitement.
FAQ
1. Qu’est-ce que la cardiomyopathie hypertrophique ?
La cardiomyopathie hypertrophique (CMH) est une maladie dans laquelle le muscle cardiaque (généralement la cloison interventriculaire) devient anormalement épaissi. Contrairement à l’hypertrophie secondaire à l’hypertension ou aux valvulopathies, dans la CMH l’épaississement est lié à des mutations génétiques et non à une surcharge du cœur.
2. La CMH est-elle héréditaire ? Faut-il examiner les apparentés ?
Oui, dans la majorité des cas la CMH est transmise selon un mode autosomique dominant. Le dépistage des apparentés au 1er degré est recommandé.
3. Quels symptômes peuvent faire suspecter une CMH et quand consulter ?
Dyspnée, douleur thoracique, vertiges, syncopes surtout à l’effort. En présence de ces plaintes ou d’antécédents familiaux de mort subite, consulter un cardiologue.
4. La CMH est-elle dangereuse ? Peut-on vivre longtemps avec ?
Avec un diagnostic précoce et un traitement adapté, la majorité des patients mènent une vie normale. Néanmoins, sans prise en charge, la CMH augmente le risque d’arythmies et de mort subite.
5. Quelle différence entre la forme obstructive et non obstructive de la CMH ?
Dans la forme obstructive, la cloison épaissie gêne l’éjection du sang du ventricule gauche, entraînant des symptômes plus marqués. Dans la forme non obstructive, l’éjection n’est pas entravée.
6. Peut-on pratiquer un sport avec une CMH ?
Les sports intenses et de compétition sont contre-indiqués. Une activité physique modérée peut être autorisée après avis médical.
7. Quels examens sont nécessaires pour poser le diagnostic de CMH ?
ECG, échocardiographie (échographie cardiaque), IRM cardiaque, Holter-ECG et test génétique (selon indications).
8. Comment traite-t-on la CMH : médicaments suffisent-ils ou faut-il opérer ?
Le traitement de première intention repose sur les médicaments. Dans les cas sévères, une intervention chirurgicale ou une ablation septale percutanée peut être nécessaire.
9. Qu’est-ce qu’un DAI et quand est-il implanté dans la CMH ?
Le DAI (défibrillateur automatique implantable) est un dispositif qui prévient la mort subite par arythmies malignes. Il est implanté chez les patients à haut risque selon les recommandations du médecin.
10. Peut-on guérir complètement la CMH ? Quel est le pronostic ?
La CMH n’est pas curable, mais les symptômes peuvent être efficacement contrôlés. Avec une prise en charge adaptée, le pronostic est favorable, surtout en l’absence de complications sévères.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy.[Prise en charge de la cardiomyopathie hypertrophique]. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management.[Cardiomyopathie hypertrophique : aperçu de la génétique et de la prise en charge]. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review.[Diagnostic et évaluation de la cardiomyopathie hypertrophique : revue JACC d’état de l’art]. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi : 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy.[Biomarqueurs circulants dans la cardiomyopathie hypertrophique] J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art.[Cardiomyopathie hypertrophique : état de l’art]. Mayo Clin Proc. 2025 Mar;100(3):557-566. doi : 10.1016/j.mayocp.2024.07.013.
St. Petersburg FL 33702, 7901 4th St N STE 300, États-Unis
Merci de votre attention !
Votre message est envoyé ! Nos experts vous contacteront dans les plus brefs délais. Si vous avez d'autres questions, n'hésitez pas à nous contacter à l'adresse suivante : info@voka.io