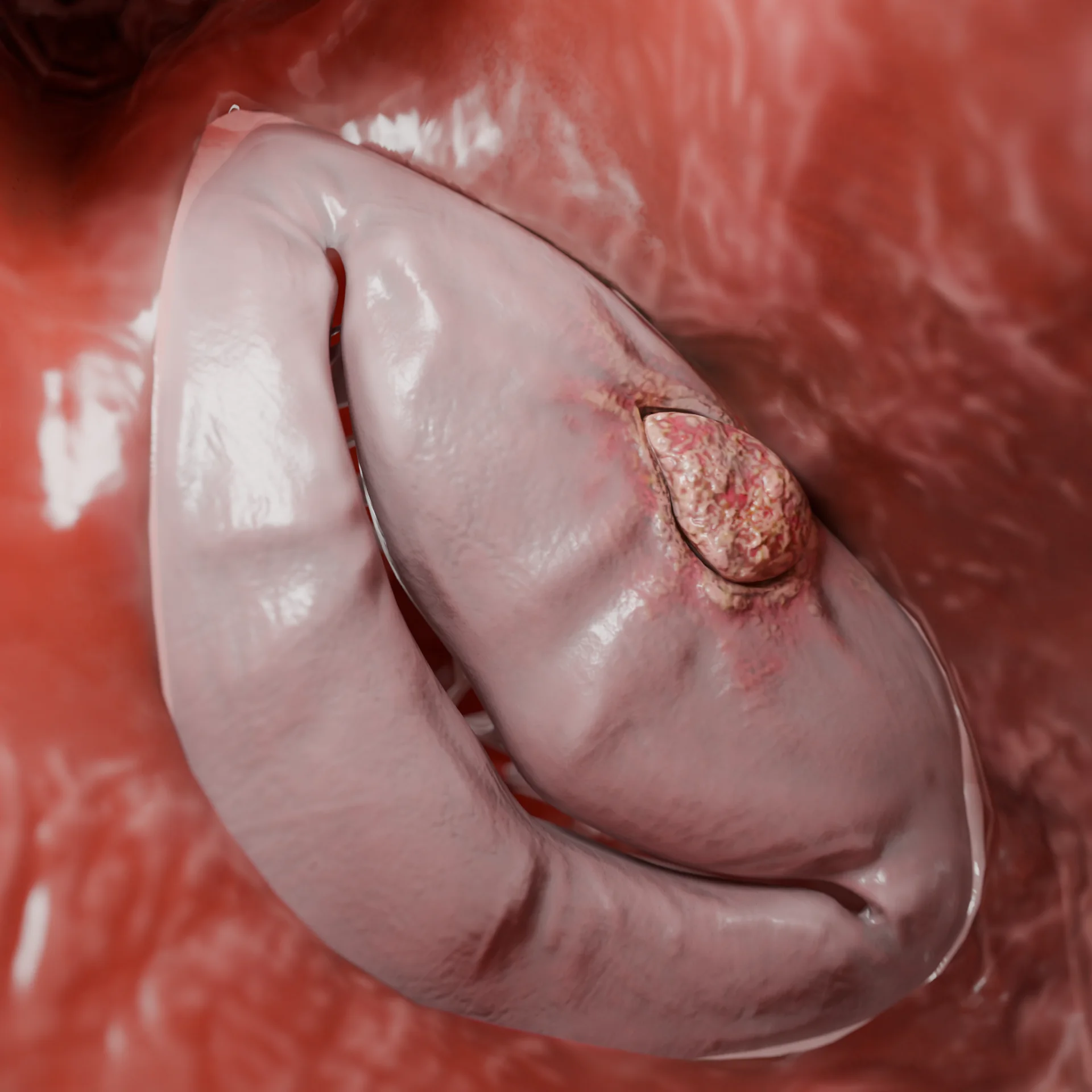
Cardiomyopathie dilatée : étiologie, pathogénie, symptômes, diagnostic, méthodes de traitement
Kizyukevich O.Chirurgien cardiovasculaire, MD
14 minutes de lecture·novembre 13, 2025
Cet article est uniquement destiné à des fins d'information
Le contenu de ce site web, y compris les textes, les graphiques et autres matériels, est fourni à titre d'information uniquement. Il ne s'agit pas d'un avis ou d'un conseil. En ce qui concerne votre état de santé ou votre traitement spécifique, veuillez consulter votre fournisseur de soins de santé.
La cardiomyopathie dilatée (CMD) est une maladie du myocarde caractérisée par une dilatation (élargissement) et un dysfonctionnement systolique du ventricule gauche ou des deux ventricules en l’absence de maladie coronarienne, de malformations congénitales, d’hypertension et d’anomalies valvulaires qui pourraient expliquer ces changements. Selon des études basées sur la population, la prévalence de la CMD est d’environ 0,036-0,400 %.
Animation 3D : cardiomyopathie dilatée
Étiologie
L’étiologie de la cardiomyopathie dilatée (CMD) est très hétérogène et comprend des causes héréditaires (génétiques/familiales) et acquises :
Causes génétiques
Elle est transmise de façon prédominante sur un mode autosomique.
Jusqu’à 50 % des cas peuvent être de nature familiale.
Les principaux gènes sont TTN, LMNA, FLNC, BAG3, DSP, RBM20, MYH7, SCN5A.
Elle peut être associée à des arythmies, à des anomalies de conduction ou à un syndrome de chevauchement (par exemple, avec des preuves de CM arythmogène).
Causes inflammatoires (post-myocardite)
Souvent secondaire à une infection virale (parvovirus B19, HHV-6, adénovirus, entérovirus).
Elle peut être auto-immune, associée à des maladies telles que le lupus érythémateux disséminé, la sarcoïdose, la polyarthrite rhumatoïde, etc.
Expositions toxiques
L’alcool a une toxicité directe sur le myocarde, en particulier à des doses élevées et en cas de consommation prolongée.
Chimiothérapie : anthracyclines (doxorubicine), trastuzumab, inhibiteurs de points de contrôle immunitaire.
La cocaïne et les amphétamines provoquent des vasospasmes et des lésions directes du myocarde.
Troubles du métabolisme
Carence en thiamine, carnitine, sélénium, zinc et cuivre.
Maladies d’accumulation : hémochromatose, maladie de Fabry, amyloïdose.
Tachyarythmie
Fibrillation auriculaire prolongée non traitée, tachycardie auriculaire ou ventriculaire, tachycardie paroxystique.
Peut être réversible avec un contrôle de la FC/du rythme cardiaque.
Cardiomyopathie du péripartum
Survient au cours des derniers mois de la grossesse ou dans les 5 mois après l’accouchement.
Cas idiopathiques
Un diagnostic d’exclusion lorsqu’aucune cause secondaire n’est identifiée et que les tests génétiques ne donnent pas d’informations.
Pathogenèse de la cardiomyopathie dilatée
Quelle qu’en soit la cause, les mécanismes pathogéniques sont similaires : lésions des cardiomyocytes, activation d’une inflammation, remodelage du myocarde et détérioration progressive de la fonction contractile.
Stades de la cardiomyopathie dilatée :
Lésions primaires des cardiomyocytes conduisant à l’activation d’une inflammation :
Génétique : perturbations de la structure du sarcomère, du noyau, du cytosquelette, etc. ;
Toxique : accumulation de radicaux libres, dysfonctionnement mitochondrial, dommages directs aux membranes des cardiomyocytes ;
Virale : inflammation chronique et fibrose résultant d’une myocardite auto-immune.
Perturbation du métabolisme du calcium intracellulaire, du métabolisme énergétique et de l’apoptose des cardiomyocytes.
Remodelage du myocarde : amincissement de la paroi, dilatation de la cavité (principalement du ventricule gauche, plus rarement des deux ventricules), développement d’une fibrose interstitielle. La dilatation annulaire et le dysfonctionnement du muscle papillaire entraînent souvent une insuffisance de la valve atrio-ventriculaire.
Dysfonctionnement systolique progressif → diminution de la fraction d’éjection.
L’activation compensatoire des systèmes neurohormonaux (SRAA, sympatho-adrénaline), qui améliore l’hémodynamique à court terme, peut conduire à terme à une insuffisance cardiaque congestive.
Paroi ventriculaire gauche fine et dilatation de la cavité : modèle 3DInsuffisance mitrale due à la dilatation de la cavité ventriculaire gauche et de l’anneau valvulaire : modèle 3D
Manifestations cliniques
Les symptômes de la cardiomyopathie dilatée sont dus à un dysfonctionnement systolique progressif et à une stase dans la circulation systémique et pulmonaire :
Dyspnée à l’effort, se transformant éventuellement en dyspnée au repos ;
Fatigue, diminution de la tolérance à l’activité physique ;
Orthopnée et dyspnée paroxystique nocturne ;
Gonflement des extrémités inférieures ;
Hypertrophie du foie, ascite ;
Syncope, vertiges (possibles en cas d’arythmie ou d’hypotension) ;
Tachycardie, troubles du rythme cardiaque (en particulier fibrillation auriculaire et arythmie ventriculaire) ;
Plus rarement, douleur thoracique due à une ischémie sous-endocardique.
Diagnostic de la cardiomyopathie dilatée
Le diagnostic de la CMD repose sur la constatation d’une dilatation du ventricule gauche et d’un dysfonctionnement systolique non expliqués par l’ischémie, l’hypertension, des malformations valvulaires ou une pathologie congénitale. L’objectif du diagnostic est de confirmer le phénotype cardiomyopathique, de déterminer les causes et d’évaluer la gravité des modifications des structures cardiaques, ainsi que la probabilité de développer des effets indésirables.
Méthodes instrumentales
L’échographie cardiaque est la principale méthode de diagnostic primaire :
Le LVEDV est augmenté (> 150-180 ml ou indexé > 75 ml/m²) ;
Fraction d’éjection réduite (< 45 %) ;
Hypokinésie globale sans anomalies régionales ;
Souvent : régurgitation mitrale et tricuspidienne, hypertension pulmonaire, dilatation du ventricule droit.
IRM cardiaque avec gadolinium (contraste tardif au gadolinium) :
Clarifie la structure du myocarde et les caractéristiques des tissus : fibrose, œdème, infiltration graisseuse ;
Schéma typique de la CMD : accumulation méso- ou sous-épicardique de gadolinium dans la paroi latérale ou septale ;
Indispensable en cas de suspicion de myocardite, de sarcoïdose et de maladies d’accumulation.
Coronarographie par tomodensitométrie/coronarographie invasive :
Elle est effectuée pour exclure une maladie coronarienne chez les patients âgés de plus de 35 ans ou en présence de facteurs de risque ;
Obligatoire en cas de douleur thoracique classique, d’anomalies régionales à l’échographie cardiaque ou d’accumulation tardive de gadolinium.
Moniteur Holter ou ECG ambulatoire (24-72 h) :
Arythmies ventriculaires (VE, VT), FA, tachycardie, pause, bloc (surtout en cas de suspicion de forme mutante LMNA) ;
Aide à la sélection des thérapies et au choix d’un DCI.
Biopsie myocardique (comme indiqué) :
En cas de suspicion : myocardite active, maladies infiltrantes (amyloïdose, sarcoïdose) ;
Utilisation limitée, nécessite des lectures précises et un haut niveau d’exécution.
Méthodes en laboratoire
BNP/NT-proBNP :
Il s’agit du biomarqueur le plus sensible de l’insuffisance cardiaque congestive. Les niveaux augmentent proportionnellement au degré de surcharge de volume et de pression. Des valeurs élevées indiquent une décompensation, des valeurs faibles permettent d’exclure l’HF.
Troponines spécifiques du cœur (I ou T) :
Une élévation modérée est possible en cas d’inflammation active (par exemple, myocardite) ou de distension myocardique marquée. Une élévation significative et aiguë nécessite l’exclusion de l’infarctus du myocarde.
Hormones thyroïdiennes (TTG, T3 et T4 libres) :
L’hypothyroïdie peut provoquer un dysfonctionnement systolique ; l’hyperthyroïdie peut provoquer une CMD induite par la tachycardie.
Glucose et hémoglobine glyquée (HbA1c) :
Le diabète sucré est associé au développement de la cardiomyopathie diabétique et exacerbe également l’évolution de l’HF.
Ferritine, fer sérique, transferrine, saturation de la transferrine :
Permettre la détection d’une carence en fer ou d’une hémochromatose. Cette dernière peut conduire à une cardiomyopathie secondaire avec dysfonctionnement progressif du ventricule gauche.
réalisée lorsqu’une nature auto-immune ou inflammatoire systémique de la maladie est suspectée : lupus érythémateux disséminé, sclérodermie, myocardite, etc.
Enzyme de conversion de l’angiotensine, récepteur soluble de l’interleukine-2 et calcium :
En cas de suspicion de sarcoïdose cardiaque. Particulièrement pertinent lorsqu’il est associé à des anomalies conductives ou à des changements infiltratifs peu clairs à l’IRM.
Tests hépatiques, créatinine, électrolytes :
Tests réguliers pour déterminer les manifestations systémiques de l’insuffisance cardiaque et pour évaluer la tolérance au traitement.
Tests génétiques :
indiqués en cas d’antécédents familiaux de cardiomyopathie, de mort subite d’origine cardiaque, de blocs, de dysfonctionnement grave à un jeune âge ou en l’absence de causes secondaires. Des panels de gènes associés à la CMD (le plus souvent TTN, LMNA, BAG3, BAG3, FLNC, SCN5A, etc.) sont utilisés.
Retrouvez d’autres contenus scientifiquement exacts sur nos médias sociaux
Abonnez-vous et ne manquez pas les dernières ressources
Traitement de la cardiomyopathie dilatée
Thérapie médicale
Le traitement médicamenteuxde la CMD nécessite une approche globale et strictement individuelle, en tenant compte des caractéristiques cliniques et fonctionnelles du patient.
Les principaux groupes de médicaments sont les suivants :
Inhibiteurs de l’ECA/ARB/ARNI (sacubitril/valsartan) : amélioration de la survie, réduction des hospitalisations ;
Bêta-bloquants (bisoprolol, carvédilol, nébivolol) : réduction de la mortalité ;
Antagonistes du récepteur minéralocorticoïde : lorsque FE < 35 % ;
Inhibiteurs du SGLT2 (dapa/empagliflozine) : amélioration du pronostic indépendamment de la présence d’un diabète ;
Diurétiques : pour les symptômes de rétention d’eau ;
Ivabradine : lorsque la FC est > 70 en rythme sinusal si les bêta-bloquants sont inadéquats ;
Anticoagulants : en cas de FA, de présence de caillots sanguins, de FC élevée.
Thérapie chirurgicale
Implantation d’un appareil :
DAI (défibrillateur automatique implantable) : si FE < 35 %, NYHA II-III et risque de VT ;
CRT-P/CRT-D (thérapie de resynchronisation) : si QRS >130 ms, FE <35 %, rythme sinusal.
Correction chirurgicale de la régurgitation mitrale (secondaire) en cas de :
Régurgitation mitrale fonctionnelle de niveau II à III ;
FEVG 30-50 %, DCD LV < 70 mm (pas de dilatation LV significative) ;
Présence de symptômes d’HF malgré un traitement médicamenteux ;
Techniques : annuloplastie (réduction de l’anneau fibreux), reconstruction par lambeau, dans certains cas MitraClip (par correction par cathéter).
Candidats à une transplantation cardiaque (en tant que pont vers la transplantation cardiaque) ;
Patients qui ne sont pas éligibles à une transplantation (dans le cadre d’un traitement de soutien à long terme).
Transplantation cardiaque :
HF réfractaire sévère (NYHA IIIb-IV) ;
Inefficacité des médicaments et des dispositifs thérapeutiques ;
Déclin progressif de la fonction des organes cibles ;
Âge généralement inférieur à 65 ans, pas de contre-indications absolues ;
Contre-indications : tumeurs malignes (avec mauvais pronostic d’espérance de vie), infections actives, hypertension pulmonaire sévère, troubles de la compliance.
FAQ
1. La cardiomyopathie dilatée peut-elle être complètement guérie ?
Pas complètement, mais avec un traitement approprié, il est possible d’améliorer de manière significative la qualité et la durée de vie.
2. Quelle est la différence entre les formes idiopathiques et génétiques de la CMD ?
La maladie idiopathique n’a pas de cause identifiée ; la maladie génétique est causée par des mutations héréditaires.
3. Pourquoi la CMD peut-elle se développer chez des personnes ne souffrant pas de maladie cardiaque ?
Elle peut être causée par des gènes cachés, des virus, des toxines, des troubles hormonaux ou une surcharge.
4. Dans quelle mesure la CMD peut-elle être mortelle et quelles sont ses principales complications ?
La cardiomyopathie dilatée est une maladie potentiellement mortelle qui évolue en l’absence de traitement, mais un traitement opportun peut réduire considérablement les risques. Son danger provient du développement de trois complications clés : l’insuffisance cardiaque progressive, qui entraîne un dysfonctionnement de plusieurs organes ; les arythmies ventriculaires malignes, qui provoquent une mort cardiaque subite ; et les événements thromboemboliques, qui peuvent conduire à un accident vasculaire cérébral mortel.
Oui, jusqu’à 50 % des cas présentent une forme familiale. Un ECG et une échographie cardiaque sont recommandés pour les proches.
7. Que signifie « fraction d’éjection réduite » ?
Il s’agit d’un indicateur de la fonction de pompage du cœur. Dans la CMD, elle est réduite en raison de l’affaiblissement du muscle cardiaque.
8. Quand un défibrillateur (DAI) doit-il être installé dans le cas d’une CMD ?
En cas de réduction sévère de FE (<35 %) et de risque d’arythmie sévère.
9. Peut-on faire du sport en cas de CMD ?
Seul un exercice modéré, convenu avec un cardiologue, est autorisé.
10. Peut-on tomber enceinte en cas de CMD ?
C’est possible, mais dans un état stable et sous surveillance médicale stricte ; les risques dépendent de la gravité de la maladie.
11. Quelles sont les caractéristiques de la cardiomyopathie dilatée chez l’enfant ?
Chez les enfants, la CMD est plus souvent associée à une myocardite antérieure ou à des syndromes génétiques spécifiques. La présentation clinique peut être non spécifique (dyspnée, difficultés d’alimentation) et le pronostic est souvent plus grave que chez l’adulte.
Références
1.
Catalogue VOKA. [Ressource électronique]
https://catalog.voka.io/
2.
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies (Lignes directrices de l’ESC pour la prise en charge des cardiomyopathies). Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi : 10.1093/eurheartj/ehad194.
3.
Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches (Cardiomyopathie dilatée : causes, mécanismes et approches thérapeutiques actuelles et futures). Lancet. 2023 Sep 16;402(10406):998-1011. doi : 10.1016/S0140-6736(23)01241-2.
4.
Gigli M, Stolfo D, Merlo M, et al. Pathophysiology of dilated cardiomyopathy: from mechanisms to precision medicine (Physiopathologie de la cardiomyopathie dilatée : des mécanismes à la médecine de précision). Nat Rev Cardiol. 2025 Mar;22(3):183-198. doi : 10.1038/s41569-024-01074-2.
5.
Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature (Cardiomyopathie dilatée : des phénotypes épidémiologiques aux phénotypes génétiques, une revue translationnelle de la littérature actuelle). J Intern Med. 2019 Oct;286(4):362-372. doi : 10.1111/joim.12944.
6.
Peters S, Johnson R, Birch S, et al. Familial Dilated Cardiomyopathy (Cardiomyopathie dilatée familiale). Heart Lung Circ. 2020 Apr;29(4):566-574. doi : 10.1016/j.hlc.2019.11.018.
7.
Harding D, Chong MHA, Lahoti N, et al. Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy (Cardiomyopathie dilatée et inflammation cardiaque chronique : pathogenèse, diagnostic et thérapie). J Intern Med. 2023 Jan;293(1):23-47. doi : 10.1111/joim.13556.
St. Petersburg FL 33702, 7901 4th St N STE 300, États-Unis
Merci de votre attention !
Votre message est envoyé ! Nos experts vous contacteront dans les plus brefs délais. Si vous avez d'autres questions, n'hésitez pas à nous contacter à l'adresse suivante : info@voka.io