Miocardiopatía hipertrófica: etiología, patogenia, síntomas, diagnóstico, métodos de tratamiento
Kizyukevich O.Cirujano cardiovascular, MD
14 min leer·julio 22, 2025
Este artículo sólo tiene fines informativos
El contenido de este sitio web, incluidos textos, gráficos y otros materiales, se proporciona únicamente con fines informativos. No pretende ser un consejo u orientación. En relación con tu enfermedad o tratamiento específico, consulta a tu médico.
La miocardiopatía hipertrófica (MCH) es una enfermedad miocárdica primaria caracterizada por una hipertrofia inexplicable de la pared del ventrículo izquierdo, generalmente asimétrica y sin dilatación de la cavidad. La enfermedad se basa en mutaciones en los genes que codifican las proteínas del sarcómero, las cuales conducen a alteraciones morfológicas, eléctricas y hemodinámicas.
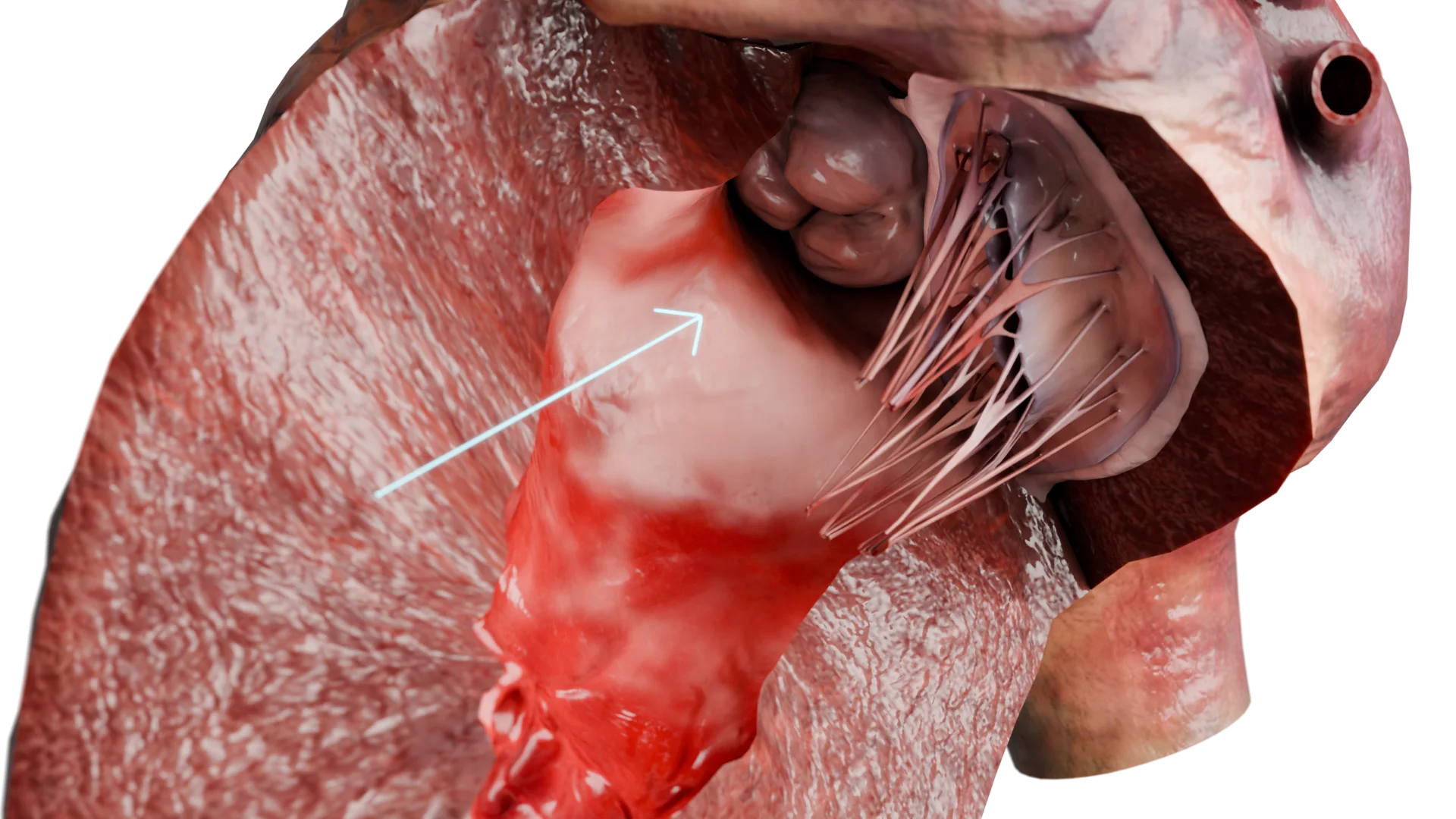
Pared del ventrículo izquierdo engrosada en la MCH: modelo 3D
La MCH es una de las formas hereditarias más frecuentes de miocardiopatía, con una prevalencia estimada de aproximadamente 1 caso por cada 500 adultos. Afecta con mayor frecuencia a los hombres que a las mujeres (en una proporción aproximada de 3:2); sin embargo, en las mujeres, la enfermedad suele diagnosticarse a una edad más avanzada y presentar un curso clínico más grave.
Etiología
La enfermedad se origina por una alteración en la síntesis y función de las proteínas contráctiles del sarcómero, pero en algunos casos se identifican formas secundarias, asociadas con trastornos sistémicos y metabólicos.
Hasta el 60–70% de los casos de miocardiopatía hipertrófica son de carácter monogénico.
Genes asociados con el desarrollo de la MCH
Gen
Proteína
Frecuencia de mutaciones
MYH7
cadena pesada de la β-miosina
Las mutaciones en MYH7 y MYBPC3 representan aproximadamente el 70% de los casos
MYBPC3
proteína C de unión a la miosina
Las mutaciones en MYH7 y MYBPC3 representan aproximadamente el 70% de los casos
TNNT2
Troponina T cardíaca
Aproximadamente el 5%
TNNI3
Troponina I
<5%
TPM1
Alfatropomiosina
<5%
La enfermedad se transmite según un patrón autosómico dominante; es decir, basta con un solo gen alterado heredado de uno de los progenitores para desarrollarla. Sin embargo, aunque la probabilidad de desarrollar la enfermedad es alta, la gravedad y la forma de presentación clínica pueden variar significativamente, incluso entre miembros de una misma familia.
Aproximadamente el 30% de los casos se deben a mutaciones de novo, sin antecedentes familiares.
Algunas enfermedades pueden imitar el cuadro clínico de la miocardiopatía hipertrófica, aunque tienen un origen patogénico diferente. Es crucial diferenciarlas de la forma primaria (sarcomérica), ya que el tratamiento y el pronóstico son distintos.
Variantes con hipertrofia secundaria (fenocopias de la MCH)
Enfermedad
Mecanismo
Características distintivas
Enfermedad de Fabry
Enfermedad por depósito lisosomal hereditaria (deficiencia de la enzima α-galactosidasa A)
Signos de afectación sistémica (angioqueratomas, neuropatía, proteinuria)
Amiloidosis cardíaca
Depósito de proteínas amiloides (AL, ATTR) en el miocardio
Enfermedad autosómica recesiva causada por mutaciones en el gen FXN (enfermedad mitocondrial)
Neurodegeneración progresiva (ataxia, debilidad y atrofia muscular, trastornos del habla, etc.)
Glucogenosis (p. ej., enfermedad de Pompe)
Acumulación de glucógeno en los lisosomas de las células, especialmente musculares y cardíacas
A menudo con afectación de la musculatura esquelética
Hipertensión arterial sistémica
Hipertrofia miocárdica reactiva
Generalmente es simétrica, con antecedentes de hipertensión
Даже при наличии мутации в гене саркомерного белка клиническая выраженность и течение ГКМП зависят от дополнительных факторов:
Reguladores epigenéticos;
Hipertensión arterial concomitante;
Actividad física de alta intensidad (especialmente durante la adolescencia);
Sexo y estado hormonal (las mujeres presentan mayor frecuencia de formas obstructivas, aunque con manifestación más tardía);
Antecedentes familiares de muerte súbita cardíaca.
Patogénesis
Trastorno de la función sarcomérica
Las mutaciones genéticas (ver etiología) provocan:
Hipersensibilidad al calcio;
Disminución de la eficiencia contráctil;
Aumento de la demanda energética.
Consecuencia: se desarrolla una hipertrofia miocárdica compensatoria, predominantemente del tabique interventricular, especialmente en la región del tracto de salida del ventrículo izquierdo (TSVI).
Hipertrofia y alteración de la relajación (disfunción diastólica)
El engrosamiento parietal conduce a:
Disminución de la distensibilidad del ventrículo izquierdo;
Alteración de su llenado en diástole;
Aumento de la presión diastólica.
Consecuencia: desarrollo de congestión en la circulación pulmonar, que se manifiesta como disnea y otros síntomas de insuficiencia cardíaca con fracción de eyección preservada.
Estrechamiento significativo del tracto de salida del VI (forma obstructiva)
En el 60–70% de los pacientes se forma una obstrucción del TSVI.
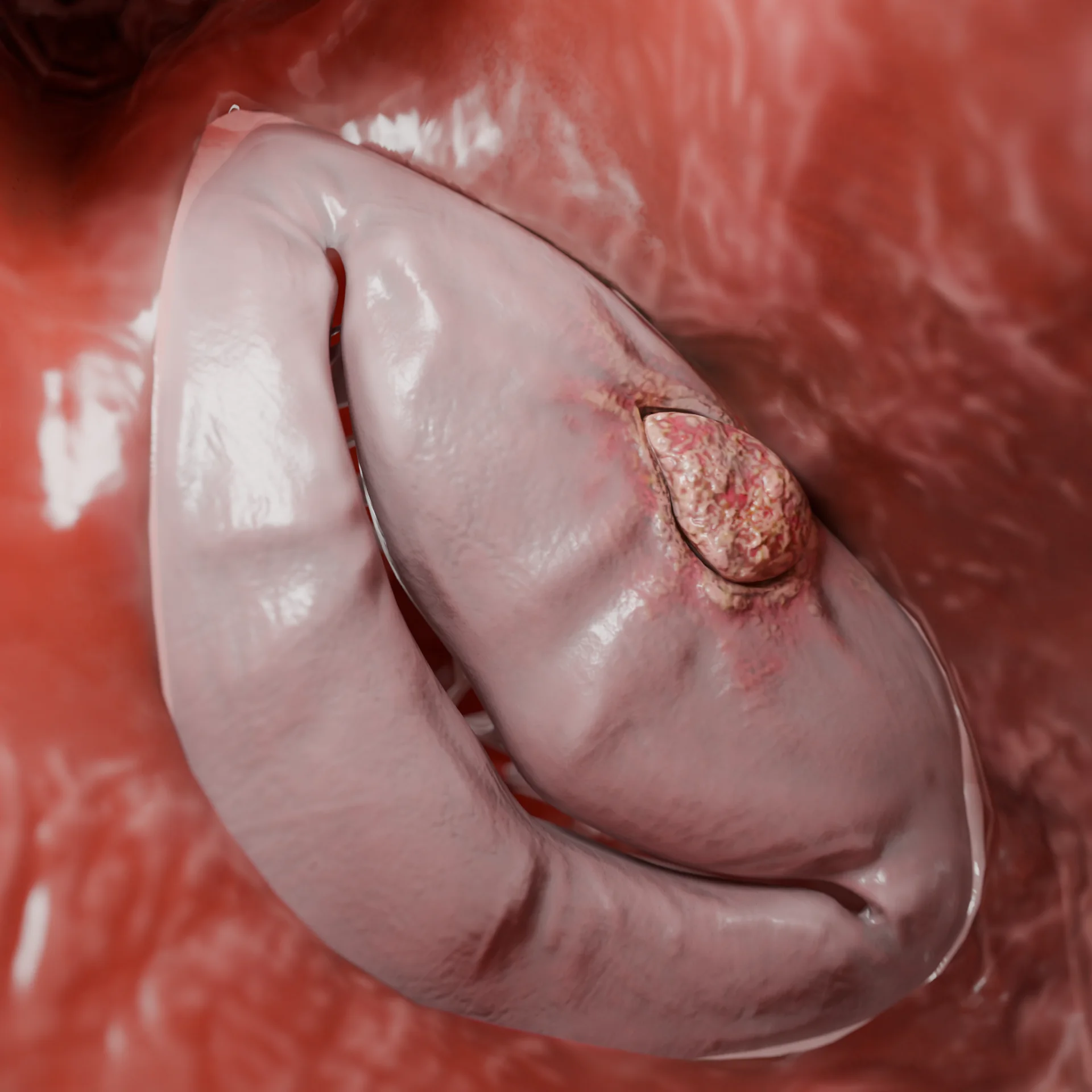
Debido al efecto Venturi durante la sístole, ocurre: desplazamiento de la valva anterior de la válvula mitral hacia el tabique (fenómeno SAM), incremento del gradiente de presión y desarrollo de regurgitación mitral.
Consecuencia: incrementa la sobrecarga hemodinámica, lo que exacerba los síntomas y eleva el riesgo de arritmias.
Obstrucción del TSVI debido al engrosamiento del tabique interventricular: modelo 3D
Isquemia microvascular
El miocardio hipertrofiado requiere más oxígeno, pero:
La red capilar no tiene tiempo de compensar el crecimiento del tejido;
Se observa un desequilibrio entre la demanda y el suministro de oxígeno;
A menudo se detecta fibrosis de los vasos de pequeño calibre.
Consecuencia: se desarrolla isquemia miocárdica a pesar de arterias coronarias normales, lo que provoca dolor torácico, fibrosis y mayor riesgo de arritmias.
Fibrosis e inestabilidad eléctrica
En respuesta a la isquemia y a la sobrecarga mecánica del miocardio, se forma fibrosis intersticial y focal, lo que conduce a:
Alteración de la conducción del impulso eléctrico;
Desarrollo de arritmias ventriculares;
Aumento del riesgo de muerte súbita cardíaca (MSC).
Animación 3D – miocardiopatía hipertrófica
Manifestaciones clínicas
Disnea de esfuerzo, fatiga;
Dolor torácico (angina de pecho) en ausencia de enfermedad coronaria;
Síncope o presíncope (especialmente durante el esfuerzo físico);
Arritmias ventriculares, fibrilación auricular;
MSC, especialmente en pacientes jóvenes y deportistas con la forma obstructiva;
Insuficiencia cardíaca: puede presentarse con fracción de eyección preservada (IC-FEp) o, en estadios avanzados, con fracción reducida. Se manifiesta con edemas, ortopnea, taquicardia, disminución de la tolerancia al ejercicio;
Curso asintomático: en el 25–30% de los pacientes, la enfermedad se detecta mediante el cribado de antecedentes familiares. No excluye un alto riesgo de complicaciones.
Diagnóstico de la miocardiopatía hipertrófica
La ecocardiografía transtorácica (ETT) es el método clave del diagnóstico inicial. Permite:
Evaluar el grosor del miocardio. El diagnóstico es probable con un grosor de pared ≥15 mm en adultos o ≥13 mm en familiares de primer grado con MCH confirmada;
Determinar la distribución de la hipertrofia: asimétrica, concéntrica o apical;
Detectar obstrucción del tracto de salida del VI (gradiente ≥30 mmHg, clínicamente significativo ≥50 mmHg);
Detectar el fenómeno SAM (movimiento sistólico anterior de la valva mitral) y la regurgitación mitral;
Evaluar la función del VI y la presencia de disfunción diastólica;
Medir las dimensiones auriculares (especialmente la aurícula izquierda, importante para el riesgo de FA).
La resonancia magnética cardíaca (RMC) se recomienda si:
La ecocardiografía no evalúa con precisión el grosor de las paredes;
Se sospecha una forma apical o atípica de MCH;
Es necesario evaluar la fibrosis miocárdica.
Identifica:
La distribución y el grado de la hipertrofia;
Áreas de fibrosis, asociadas con un mayor riesgo de arritmias y MSC;
La diferenciación con fenocopias (por ejemplo, amiloidosis).
El electrocardiograma (ECG) es inespecífico, pero muestra cambios patológicos en más del 90% de los pacientes. Los signos de hipertrofia ventricular izquierda incluyen:
Ondas Q atípicas (en las derivaciones V4–V6, I, aVL), las cuales pueden simular un infarto de miocardio;
Alteraciones del segmento ST e inversión de la onda T;
Trastornos del ritmo: fibrilación auricular, extrasístoles ventriculares, taquicardia ventricular;
Bloqueos auriculoventriculares o intraventriculares.
Monitorización con Holter. Indicaciones:
Sospecha de arritmias (extrasístoles ventriculares, taquicardia ventricular, fibrilación auricular);
Síncope o presíncope;
Evaluación de la gravedad de los trastornos del ritmo e indicaciones de DAI.
Prueba de esfuerzo (ergometría en tapiz rodante o cicloergometría). Se realiza para:
Evaluar la tolerancia al ejercicio físico;
Detectar obstrucción inducible del TSVI;
Determinar el gradiente durante el esfuerzo;
Identificar síntomas isquémicos en ausencia de estenosis de las arterias coronarias.
Pruebas genéticas. Se recomiendan:
A pacientes con MCH confirmada (especialmente jóvenes o con antecedentes familiares de MSC);
Para el cribado de familiares de primer grado (hijos, hermanos, padres);
En casos de sospecha de fenocopias (enfermedad de Fabry, enfermedades mitocondriales, etc.).
Genes que se detectan (los más frecuentemente asociados): MYH7, MYBPC3, TNNT2, TNNI3, TPM1.
Marcadores de laboratorio:
NT-proBNP / BNP: se elevan en casos de sobrecarga de presión y disfunción diastólica;
Troponina T/I: puede estar moderadamente elevada debido a la isquemia microvascular.
Encuentra más contenido científicamente preciso en nuestras redes sociales
Suscríbete y no te pierdas los últimos recursos
Tratamiento de la miocardiopatía hipertrófica
Modificación del estilo de vida:
Exclusión de esfuerzos físicos intensos y deportes;
Control de la presión arterial y el peso corporal;
Verapamilo: en caso de contraindicación a los β-bloqueantes;
Disopiramida: como tratamiento coadyuvante en la forma obstructiva;
Mavacamten: un fármaco nuevo con eficacia demostrada para reducir el gradiente y mejorar los síntomas (según las guías ESC 2023 y AHA 2020).
Tratamiento quirúrgico
Indicaciones:
Gradiente en el tracto de salida del ventrículo izquierdo (TSVI) ≥50 mmHg en reposo o con provocación;
Síntomas graves (NYHA III–IV) refractarios a β-bloqueantes, verapamilo o disopiramida;
Regurgitación mitral significativa asociada al fenómeno SAM.
Miectomía septal ampliada:
Es el «estándar de oro» quirúrgico para la MCH obstructiva;
Se realiza a través de miniesternotomía o esternotomía completa; en algunos casos, es posible mediante minitoracotomía anterior derecha;
Se extirpa una porción del tabique interventricular hipertrófico, eliminando el gradiente de presión;
Si es necesario, se realiza una plastia de la válvula mitral o una resección de las cuerdas secundarias;
Complicaciones potenciales: bloqueos cardíacos, necesidad de DAI, recurrencia del gradiente.
La ablación septal con alcohol (intervención endovascular) se utiliza con mayor frecuencia en pacientes para los que la cirugía abierta está contraindicada o tiene riesgos demasiado elevados.
Inyección de etanol en una arteria perforante septal → infarto localizado → reducción del grosor septal;
Riesgo de bloqueo AV completo (hasta un 10%), lo que requiere estar preparado para implantar un marcapasos;
Menor previsibilidad de los resultados;
Posibilidad de eliminación incompleta de la obstrucción.
Implantación de un desfibrilador automático implantable (DAI)
Indicaciones:
MSC recuperada o taquicardia ventricular sostenida;
Grosor de la pared del VI >30 mm;
Antecedentes familiares de MSC;
Síncope de etiología no aclarada;
FEVI <50% en casos de evolución progresiva.
Trasplante cardíaco en pacientes en estadio terminal con insuficiencia cardíaca refractaria a pesar del tratamiento.
FAQ
1. ¿Qué es la miocardiopatía hipertrófica?
La miocardiopatía hipertrófica (MCH) es una enfermedad en la que el músculo cardíaco (normalmente el tabique entre los ventrículos) se engrosa de forma anormal. A diferencia de la hipertrofia debida a la hipertensión o a defectos valvulares, en la MCH el engrosamiento se debe a mutaciones genéticas y no a una sobrecarga del corazón.
2. ¿Es hereditaria la MCH? ¿Es necesario examinar a los familiares?
Sí, en la mayoría de los casos, la MCH se hereda con un patrón autosómico dominante. Se recomienda examinar a los familiares de primer grado.
3. ¿Qué síntomas pueden indicar MCH y cuándo hay que acudir al médico?
Los síntomas incluyen síncope, dolor torácico, mareos, desmayos, sobre todo al hacer esfuerzos. Si presenta estos síntomas o hay antecedentes familiares de muerte súbita, debe consultar a un cardiólogo.
4. ¿Es peligrosa la MCH? ¿Se puede vivir mucho tiempo con ella?
Con un diagnóstico oportuno y un tratamiento adecuado, la mayoría de los pacientes llevan una vida plena. Sin embargo, en algunos casos, la MCH aumenta el riesgo de arritmias y muerte súbita, sobre todo sin tratamiento.
5. ¿En qué se diferencia la MCH obstructiva de la MCH no obstructiva?
En la forma obstructiva, el tabique engrosado obstruye el flujo de salida de sangre del ventrículo izquierdo, lo que provoca síntomas más intensos. En la forma no obstructiva, el flujo de salida no está obstruido.
6. ¿Se puede hacer deporte teniendo MCH?
Los deportes intensos y competitivos no están recomendados a las personas con MCH. Se permite una actividad física moderada, siempre consensuada con el médico tratante.
7. ¿Qué exámenes son necesarios para diagnosticar la MCH?
Suelen realizarse: electrocardiograma, ecocardiograma (ecocardiografía), resonancia magnética cardíaca, monitorización ambulatoria del electrocardiograma (Holter) y, si está indicado, pruebas genéticas.
8. ¿Cómo se trata la MCH: es necesaria la cirugía o bastan las pastillas?
La base del tratamiento son los medicamentos. En casos graves, puede requerirse cirugía o ablación septal percutánea.
9. ¿Qué es un DAI y cuándo se coloca en la MCH?
El DAI (desfibrilador automático implantable) es un dispositivo que previene la muerte súbita por arritmias. Se coloca en pacientes con alto riesgo según las recomendaciones médicas.
10. ¿Es posible curar la MCH por completo? ¿Cuál es el pronóstico?
La MCH no tiene cura definitiva, pero sus síntomas se pueden controlar de manera eficaz. Con el manejo correcto, el pronóstico es favorable, especialmente si no se presentan complicaciones graves.
Zhang Y, Adamo M, Zou C, Porcari A, Tomasoni D, Rossi M, Merlo M, Liu H, Wang J, Zhou P, Metra M, Sinagra G, Zhang J. Management of hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2024 Jun 1;25(6):399-419. doi: 10.2459/JCM.0000000000001616.
4.
Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019 Dec 16;9(12):878. doi: 10.3390/biom9120878.
5.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022 Feb 1;79(4):372-389. doi: 10.1016/j.jacc.2021.12.002.
6.
Matthia EL, Setteducato ML, Elzeneini M, Vernace N, Salerno M, Kramer CM, Keeley EC. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2022 Dec 6;11(23):e027618. doi: 10.1161/JAHA.122.027618.
7.
Ommen SR, Nishimura RA, Schaff HV, Dearani JA. Hypertrophic Cardiomyopathy: State of the Art. Mayo Clin Proc. Marzo de 2025; 100(3):557-566. doi: 10.1016/j.mayocp.2024.07.013.
Petersburg FL 33702, 7901 4th St N STE 300, ESTADOS UNIDOS
¡Gracias!
¡Tu mensaje ha sido enviado! Nuestros expertos se pondrán en contacto contigo en breve. Si tienes más preguntas, ponte en contacto con nosotros en info@voka.io.