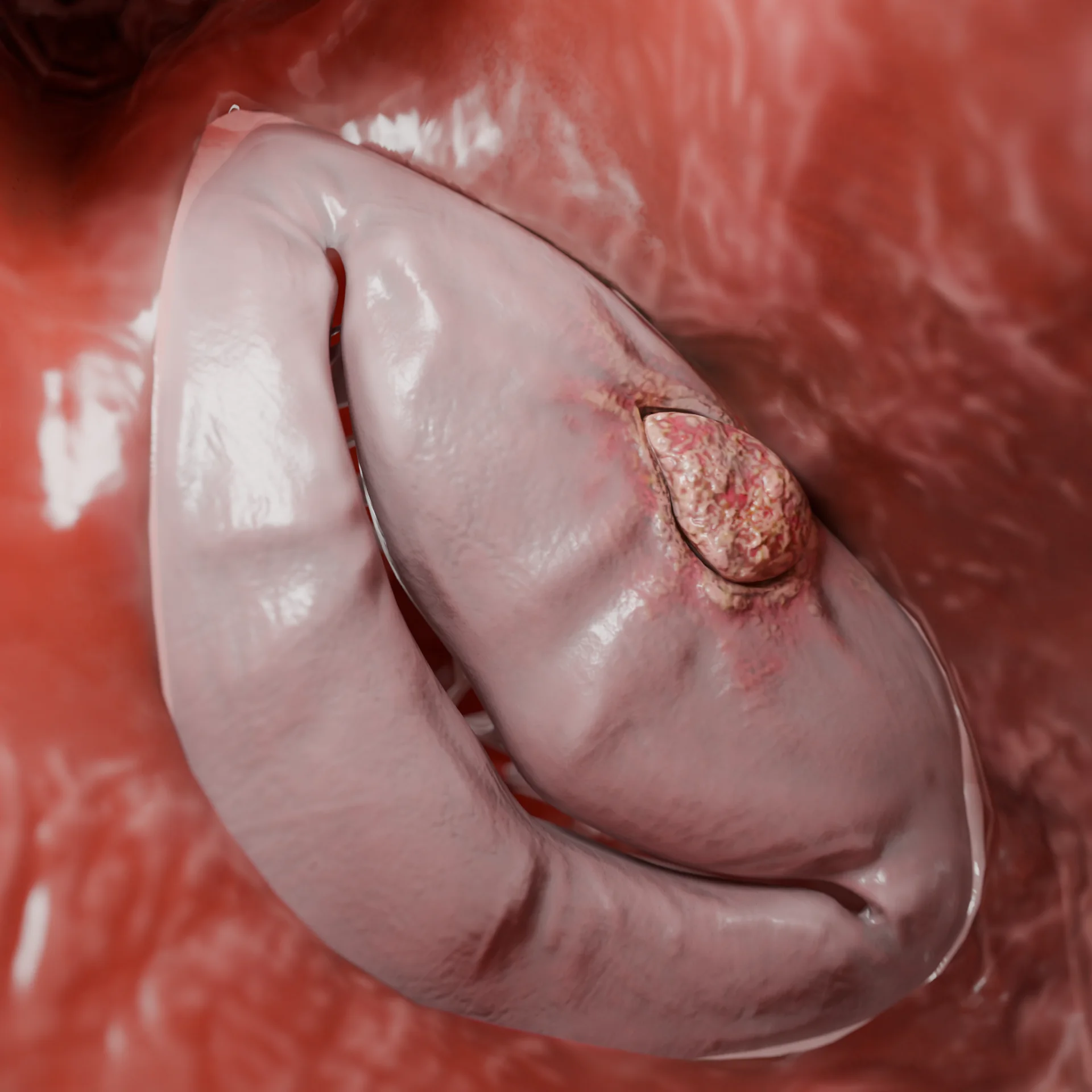
Miocardiopatía dilatada: etiología, patogénesis, síntomas, diagnóstico, métodos de tratamiento
Kizyukevich O.Cirujano cardiovascular, MD
14 min leer·noviembre 13, 2025
Este artículo sólo tiene fines informativos
El contenido de este sitio web, incluidos textos, gráficos y otros materiales, se proporciona únicamente con fines informativos. No pretende ser un consejo u orientación. En relación con tu enfermedad o tratamiento específico, consulta a tu médico.
La miocardiopatía dilatada (MCD) es una enfermedad miocárdica caracterizada por la dilatación (aumento de tamaño) y la disfunción sistólica del ventrículo izquierdo o de ambos ventrículos en ausencia de enfermedad coronaria, malformaciones congénitas, hipertensión y anomalías valvulares que pudieran explicar estos cambios. De acuerdo con los estudios poblacionales, la prevalencia de la MCD es de aproximadamente el 0,036–0,400 %.
Animación 3D: miocardiopatía dilatada
Etiología
La etiología de la miocardiopatía dilatada (MCD) es muy heterogénea e incluye causas hereditarias (genéticas/familiares) y adquiridas:
Causas genéticas
Se hereda de forma predominantemente autosómica dominante.
Hasta el 50 % de los casos pueden ser de carácter familiar.
Los genes principales son TTN, LMNA, FLNC, BAG3, DSP, RBM20, MYH7, SCN5A.
Puede estar combinada con arritmias, anomalías de la conducción o un fenotipo superpuesto (por ejemplo, con evidencia de miocardiopatía arritmogénica).
Causas inflamatorias (postmiocarditis)
Con frecuencia, la causa es una infección viral (parvovirus B19, HHV-6, adenovirus, enterovirus).
Puede ser autoinmune o estar asociada a enfermedades como el lupus eritematoso sistémico, la sarcoidosis, la artritis reumatoide, etc.
Exposiciones tóxicas
El alcohol tiene una toxicidad miocárdica directa, especialmente en dosis altas y con un uso prolongado.
Quimioterapia: antraciclinas (doxorubicina), trastuzumab, inhibidores de puntos de control inmunológico.
La cocaína y las anfetaminas provocan vasoespasmo y daño miocárdico directo.
Trastornos metabólicos
Deficiencia de tiamina, carnitina, selenio, zinc y cobre.
Enfermedades de acumulación: hemocromatosis, enfermedad de Fabry, amiloidosis.
Taquiarritmias
Fibrilación auricular prolongada no tratada, taquicardia auricular o ventricular, taquicardia paroxística.
Puede ser reversible mediante el control de la frecuencia cardíaca/ritmo cardíaco.
Miocardiopatía periparto
Se produce en los últimos meses del embarazo o en los 5 meses posteriores al parto.
Casos idiopáticos
Diagnóstico de exclusión, cuando no se identifican causas secundarias y las pruebas genéticas no aportan información.
Patogénesis de la miocardiopatía dilatada
Independientemente de la causa, los mecanismos patogénicos son similares: daño a los miocardiocitos, activación de la inflamación, remodelación miocárdica y deterioro progresivo de la función contráctil.
Etapas de la miocardiopatía dilatada:
Daño primario a los miocardiocitos que da lugar a la activación de la inflamación:
Genético: alteraciones en la estructura del sarcómero, del núcleo, del citoesqueleto, etc.;
Tóxico: acumulación de radicales libres, disfunción mitocondrial, daño directo a las membranas de los miocardiocitos;
Vírico: inflamación crónica y fibrosis como resultado de una miocarditis autoinmune.
Trastornos del metabolismo intracelular del calcio, del metabolismo energético y de la apoptosis de los miocardiocitos.
Remodelación miocárdica: adelgazamiento de la pared, dilatación de la cavidad (principalmente, del ventrículo izquierdo; con menor frecuencia, de ambos ventrículos) y desarrollo de fibrosis intersticial. Como consecuencia de la dilatación anular y la disfunción del músculo papilar, a menudo se desarrolla insuficiencia de las válvulas auriculoventriculares.
Disfunción sistólica progresiva → disminución de la fracción de eyección.
La activación compensatoria de los sistemas neurohormonales (RAAS, simpático-adrenal), que mejora la hemodinámica a corto plazo, puede provocar insuficiencia cardíaca congestiva con el tiempo.
Pared delgada del ventrículo izquierdo y dilatación de la cavidad: modelo 3DInsuficiencia mitral debida a dilatación de la cavidad del ventrículo izquierdo y del anillo valvular: modelo 3D
Manifestaciones clínicas
Los síntomas de la miocardiopatía dilatada se deben a la disfunción sistólica progresiva y a la estasis tanto en la circulación sistémica como en la pulmonar:
Disnea de esfuerzo, que acaba evolucionando a disnea en reposo;
Fatiga, disminución de la tolerancia a la actividad física;
Ortopnea y disnea paroxística nocturna;
Hinchazón de las extremidades inferiores;
Hepatomegalia, ascitis;
Síncope, mareo (es posible con arritmias o presión arterial baja);
Taquicardia, trastornos del ritmo (especialmente fibrilación auricular y arritmias ventriculares);
Con menor frecuencia, dolor torácico debido a isquemia subendocárdica.
Diagnóstico de la miocardiopatía dilatada
El diagnóstico de la MCD se basa en la presencia de dilatación del ventrículo izquierdo y disfunción sistólica que no se explica por isquemia, hipertensión, malformaciones valvulares o patología congénita. El objetivo del diagnóstico es confirmar el fenotipo cardiomiopático, determinar las causas y evaluar la gravedad de las alteraciones en las estructuras cardíacas, así como la probabilidad de desarrollar efectos adversos.
Métodos instrumentales
La ecografía cardíaca es el método principal de diagnóstico primario:
El volumen telediastólico del ventrículo izquierdo está aumentado (>150–180 ml o indexado >75 ml/m²).
Fracción de eyección reducida (<45 %).
Hipocinesia global sin anomalías regionales.
Con frecuencia: insuficiencia de las válvulas mitral y tricúspide, hipertensión pulmonar, dilatación del ventrículo derecho.
Resonancia magnética cardíaca con gadolinio (contraste tardío de gadolinio):
Aclara la estructura miocárdica y las características del tejido: fibrosis, edema, infiltración grasa.
Patrón típico en la miocardiopatía dilatada: acumulación meso o subepicárdica de gadolinio en la pared lateral o septal.
Indispensable en casos de sospecha de miocarditis, sarcoidosis y enfermedades acumulativas.
Angiografía coronaria por TC/angiografía coronaria invasiva:
Se realiza para descartar la cardiopatía isquémica en pacientes mayores de 35 años o en presencia de factores de riesgo.
Es obligatoria en casos de dolor torácico típico, anomalías regionales en la ecografía cardíaca o acumulación tardía de gadolinio.
Monitor Holter o ECG ambulatorio (24–72 h):
Arritmias ventriculares (VE, TV), FA, taquicardia, pausa, bloqueo (especialmente si se sospecha la forma mutante LMNA).
Ayuda a seleccionar las terapias y a decidir sobre la necesidad de utilizar un desfibrilador automático implantable (DAI).
Biopsia miocárdica (según se indique):
En caso de sospecha de lo siguiente: miocarditis activa, enfermedades infiltrativas (amiloidosis, sarcoidosis).
Su uso es limitado, se requieren lecturas precisas y un alto nivel de ejecución.
Métodos de laboratorio
BNP/NT-proBNP:
Es el biomarcador más sensible de la insuficiencia cardíaca congestiva. Los niveles aumentan en proporción al grado de sobrecarga de volumen y presión. Los valores altos indican descompensación, los valores bajos permiten descartar insuficiencia cardíaca.
Troponinas específicas cardíacas (I o T):
Es posible una elevación moderada con inflamación activa (por ejemplo, miocarditis) o distensión miocárdica marcada. Una elevación significativa y aguda requiere descartar un infarto de miocardio.
Hormonas tiroideas (TTG, T3 libre y T4):
El hipotiroidismo puede causar disfunción sistólica, el hipertiroidismo puede causar miocardiopatía dilatada inducida por taquicardia.
Glucosa y hemoglobina glicosilada (HbA1c):
La diabetes mellitus se asocia con el desarrollo de cardiomiopatía diabética y también exacerba el curso de la insuficiencia cardíaca.
Ferritina, hierro sérico, transferrina, saturación de transferrina:
Permite detectar deficiencia de hierro o hemocromatosis. Esto último puede producir una miocardiopatía secundaria con disfunción progresiva del ventrículo izquierdo.
Marcadores de inflamación y autoinmunidad (anticuerpos antinucleares, factor reumatoide, anticuerpos anticardiolipina, etc.):
Se utilizan cuando existen sospechas acerca de la naturaleza autoinmune o inflamatoria sistémica de la enfermedad, como lupus eritematoso sistémico, esclerodermia, miocarditis, etc.
Enzima convertidora de angiotensina, receptor soluble de interleucina-2 y calcio:
Se utiliza cuando se sospecha sarcoidosis cardíaca. Resulta de especial importancia cuando se combina con anomalías conductivas o cambios infiltrativos poco claros en la resonancia magnética.
Pruebas hepáticas, creatinina, electrolitos:
Se realizan pruebas de forma rutinaria para determinar las manifestaciones sistémicas de la insuficiencia cardíaca y evaluar la tolerabilidad del tratamiento.
Pruebas genéticas:
Están indicadas en caso de antecedentes familiares de miocardiopatía, muerte súbita cardíaca, bloqueos, disfunción grave a una edad temprana o ausencia de causas secundarias. Se utilizan paneles para genes asociados con la MCD (con mayor frecuencia, TTN, LMNA, BAG3, BAG3, FLNC, SCN5A, etc.).
Encuentra más contenido científicamente preciso en nuestras redes sociales
Suscríbete y no te pierdas los últimos recursos
Tratamiento de la miocardiopatía dilatada
Terapia farmacológica
La terapia farmacológicapara la MCD requiere un enfoque integral y estrictamente individualizado, en el que se consideren las características clínicas y funcionales de cada paciente.
Los principales grupos de fármacos son los siguientes:
Inhibidores de la ECA/BRA/ARNI (sacubitrilo/valsartán): mejoran la supervivencia y reducen las hospitalizaciones;
Betabloqueantes (bisoprolol, carvedilol, nebivolol): reducen la mortalidad;
Antagonistas mineralocorticoides: cuando la fracción de eyección es <35 %;
Inhibidores de SGLT2 (dapa/empagliflozina): mejor pronóstico independientemente de la presencia de diabetes;
Diuréticos: para los síntomas de retención de líquidos;
Ivabradina: cuando la frecuencia cardíaca es >70 en ritmo sinusal si los betabloqueantes son inadecuados;
Anticoagulantes: en caso de fibrilación auricular, presencia de coágulos sanguíneos, frecuencia cardíaca elevada.
Terapia quirúrgica
Implantación de dispositivos:
DAI (desfibrilador automático implantable): si la fracción de eyección es <35 %, clase II-III de la NYHA y existe riesgo de taquicardia ventricular.
TRC-P/TRC-D (terapia de resincronización cardíaca): si el QRS es >130 ms, la fracción de eyección es <35 % y hay ritmo sinusal.
Corrección quirúrgica de la insuficiencia de la válvula mitral (secundaria) ante lo siguiente:
Insuficiencia de la válvula mitral funcional de grado II a III;
Fracción de eyección del ventrículo izquierdo 30–50 %, diámetro telediastólico del ventrículo izquierdo <70 mm (sin dilatación significativa del ventrículo izquierdo);
Presencia de síntomas de insuficiencia cardíaca a pesar de la terapia farmacológica.
Técnicas: anuloplastia (reducción del anillo fibroso), reconstrucción con colgajo, en algunos casos MitraClip (corrección mediante catéter).
Ineficacia de la terapia con medicamentos y dispositivos;
Deterioro progresivo de la función del órgano diana;
En general, edad <65 años, sin contraindicaciones absolutas.
Contraindicaciones: tumores malignos (con mal pronóstico de supervivencia), infecciones activas, hipertensión pulmonar grave, trastornos de la conformidad.
FAQ
1. ¿La miocardiopatía dilatada tiene cura?
No del todo; sin embargo, con el tratamiento adecuado, es posible mejorar significativamente la calidad y la duración de la vida.
2. ¿Cuál es la diferencia entre las formas idiopáticas y genéticas de la MCD?
La idiopática no tiene causa identificada; la genética está causada por mutaciones heredadas.
3. ¿Por qué puede desarrollarse la MCD en personas sin enfermedad cardíaca?
Puede deberse a genes ocultos, virus, toxinas, trastornos hormonales o sobrecarga.
4. ¿En qué medida es mortal la MCD y cuáles son sus principales complicaciones?
La miocardiopatía dilatada es una enfermedad potencialmente mortal que progresa sin tratamiento, pero se pueden reducir significativamente los riesgos con una terapia oportuna. Su peligro radica en el desarrollo de tres complicaciones clave: insuficiencia cardíaca progresiva, que produce una disfunción multiorgánica; arritmias ventriculares malignas, que causan muerte súbita cardíaca; y eventos tromboembólicos, que pueden provocar un accidente cerebrovascular mortal.
5. ¿Qué síntomas deberían ser una señal de alerta?
Disnea, edema, fatiga, palpitaciones, desmayos.
6. ¿La enfermedad es hereditaria?
Sí, hasta el 50 % de los casos tiene un componente familiar. Se recomienda realizar un electrocardiograma y una ecografía cardíaca a los familiares.
7. ¿Qué significa «fracción de eyección reducida»?
Es un indicador de la función de bombeo del corazón. En la MCD, se reduce debido al debilitamiento del músculo cardíaco.
8. ¿Cuándo se debe implantar un desfibrilador (DAI) en la MCD?
En casos graves de reducción de la fracción de eyección (<35 %) y riesgo de arritmias graves.
9. ¿Se puede hacer ejercicio con MCD?
Solo se permite el ejercicio moderado, previa aprobación de un cardiólogo.
10. ¿Es posible quedar embarazada con MCD?
Es posible, pero en condiciones estables y bajo estricta supervisión médica; los riesgos dependen de la gravedad de la enfermedad.
11. ¿Cuáles son las características de la miocardiopatía dilatada en niños?
En los niños, la MCD se asocia con mayor frecuencia a antecedentes de miocarditis o síndromes genéticos específicos. La presentación clínica puede ser inespecífica (disnea, dificultades para alimentarse) y el pronóstico suele ser más grave que en los adultos.
Bibliografía
1.
Catálogo VOKA. [Recurso electrónico]
https://catalog.voka.io/
2.
Arbelo E, Protonotarios A, Gimeno JR, et al. Guías ESC 2023 para el tratamiento de las miocardiopatías [2023 ESC Guidelines for the management of cardiomyopathies]. Eur Heart J. 2023 Oct 1;44(37):3503-3626. doi: 10.1093/eurheartj/ehad194.
3.
Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Miocardiopatía dilatada: causas, mecanismos y enfoques de tratamiento actuales y futuros [Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches]. Lancet. 16 de septiembre de 2023; 402(10406):998-1011. doi: 10.1016/S0140-6736(23)01241-2.
4.
Gigli M, Stolfo D, Merlo M, et al. Fisiopatología de la miocardiopatía dilatada: de los mecanismos a la medicina de precisión [Pathophysiology of dilated cardiomyopathy: from mechanisms to precision medicine]. Nat Rev Cardiol. Marzo de 2025; 22(3):183-198. doi: 10.1038/s41569-024-01074-2.
5.
Reichart D, Magnussen C, Zeller T, Blankenberg S. Miocardiopatía dilatada: de los fenotipos epidemiológicos a los genéticos: una revisión traslacional de la literatura actual [Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature]. J Intern Med. Octubre de 2019; 286(4):362-372. doi: 10.1111/joim.12944.
6.
Peters S, Johnson R, Birch S, et al. Miocardiopatía dilatada familiar [Familial Dilated Cardiomyopathy]. Heart Lung Circ. Abril de 2020; 29(4):566-574. doi: 10.1016/j.hlc.2019.11.018.
7.
Harding D, Chong MHA, Lahoti N, et al. Miocardiopatía dilatada e inflamación cardíaca crónica: patogénesis, diagnóstico y tratamiento [Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy]. J Intern Med. Enero de 2023; 293(1):23-47. doi: 10.1111/joim.13556.
Petersburg FL 33702, 7901 4th St N STE 300, ESTADOS UNIDOS
¡Gracias!
¡Tu mensaje ha sido enviado! Nuestros expertos se pondrán en contacto contigo en breve. Si tienes más preguntas, ponte en contacto con nosotros en info@voka.io.